
Amino acid discrimination in a nanopore and the feasibility of sequencing peptides with a tandem cell and exopeptidase†
G. Sampath*
59 Washington Street #178, Santa Clara, CA 95050, USA. E-mail: sampath_2068@yahoo.com
First published on 13th March 2015
Abstract
In polymer sequencing with a nanopore multiple discriminators may be used to distinguish among more monomers than usual. This leads to a proposal to sequence peptides using a tandem cell (RSC Adv., 2015, 5, 167–171) with exopeptidase. A Fokker–Planck model of the device shows that the 20 amino acids display a well-defined ordering, and that, in theory, several necessary conditions for effective sequencing are satisfied. Other factors, such as the role of solution pH, exopeptidase cleaving efficiency, and modified amino acids, are discussed. If validated experimentally this approach could lead to an alternative to mass spectrometry.
Background
In nanopore sequencing, an analyte (polymer) translocates through a biological nanopore embedded in a bilipid membrane or a hole drilled through a synthetic membrane separating the cis and trans chambers of an electrolytic cell with an aqueous solution of KCl. A potential difference applied between the two chambers causes ionic (K+, Cl−) current flow. As the analyte passes through the pore it causes a current blockade, which is used to identify it (or its components). Alternatively a graphene sheet or layer of molybdenum sulphide with a nano-sized hole may be used with a transverse current through the sheet to identify the analyte. While studies of nanopore-based sequencing of DNA have led to practical implementations,1 in contrast sequencing of proteins or peptide strands using nanopores is still in an early stage.2 Most of this work is on protein unfolding,3 identification of whole proteins4 or domains within,5 detecting modifications such as phosphorylation,6 or conformation studies.7 A recent report describes the use of transverse electrodes and residue-specific detector molecules in the pore to measure a transverse tunneling current through a single amino acid in a translocating peptide.8This report looks at the possibility of sequencing a peptide using a modified version of a tandem electrolytic cell that was previously proposed for DNA sequencing and modeled mathematically.9 The cell has two pores in tandem and an exopeptidase attached to the downstream side of the first pore. The enzyme cleaves the leading residue from a peptide strand drawn into the first pore by a potential difference. The cleaved residue translocates through the second pore and is detected based on the current blockade. Sequence identification uses multiple discriminators such as the current blockade level (or a variant) and different translocation times. If experimentally validated this approach could lead to a feasible alternative to mass spectrometry (ESI/MALDI)10 and gel electrophoresis.11
Any attempt to sequence proteins using nanopores must consider the following: (1) amino acids that make up proteins are charged to varying degrees depending on the solution pH (see Table 2 in the ESI†); therefore the ability of a protein to move in an electric field depends on the net charge; (2) native proteins must be unfolded before sequencing can begin; (3) homopolymers, that is, successive repeats of amino acids (or residues, the two terms are used interchangeably below) in the sequence, may present a problem; (4) proteins are not easily replicated; (5) if sequencing is based on cleaving of a strand (‘exosequencing’) the original sequence is not easily reconstructed; and (6) complex algorithms based on Markov–Viterbi models or neural nets (such as those used to extract base-level information in DNA sequences from signals due to k-mers12,13) have to deal with 20 amino acid types (compared with 4 in DNA). Some of these problems can be alleviated: (1) molecules with low charge can be made more mobile with a hydraulic pressure gradient;14,15 (2) folded proteins can be unfolded using a nanopore;3 (3) the homopolymer problem can be solved in part by ‘exosequencing’, that is, individual residues are cleaved from the peptide for identification (similar to exosequencing of DNA16 with a tandem cell9); and (4) multiple discriminators can be used to distinguish among the 20 amino acid types. The tandem cell approach to peptide/protein sequencing proposed here is based on these notions.
A tandem cell for peptide sequencing
Fig. 1 shows a schematic of a modified tandem cell based on the generic form [cis1, upstream pore (UNP), trans1/cis2, downstream pore (DNP), trans2].9 An exopeptidase (amino or carboxy) attached to the downstream side of UNP cleaves the leading residue of a peptide threaded from cis1 through UNP to trans1/cis2. A potential difference V05 (normally > 0) is applied between cis1 and trans2 over the five sections; most of it (∼98%) drops across the two pores.1 Analyte translocation is primarily based on diffusion supplemented by drift due to the electric field E = Vij/Lij in each section (0 ≤ i < 5, j = i + 1). The diffusion-drift process can be modeled with a Fokker–Planck equation, and the mean and standard deviation of the times taken by the particle to translocate through a chamber (cis or trans) and through a pore of length L calculated. With the pore axis parallel to the z axis and directed from cis1 to trans2, a residue has mobility μ that varies with the effective charge (which is determined by the solution pH) and experiences a drift velocity vz that could be zero, negative, or positive, and a translocation time that is equal to, greater than, or less than that due to diffusion alone.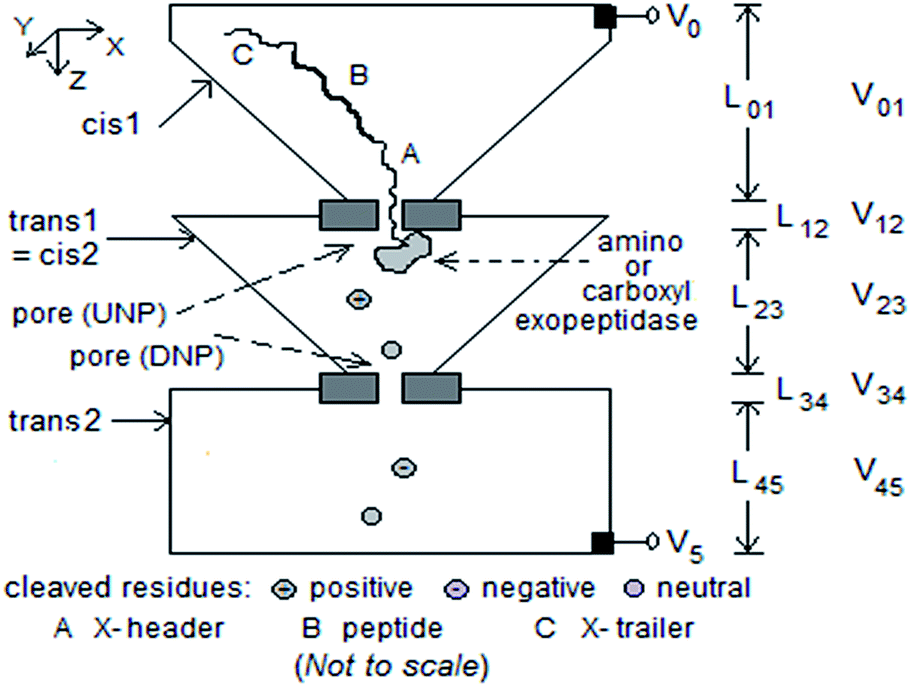 | ||
| Fig. 1 Schematic of modified tandem cell for peptide sequencing with five pipelined stages. Dimensions considered: (1) cis1: box of height 1 μm tapering to cross-section 100 nm2; (2) UNP: length 10–20 nm, diameter 10 nm; (3) trans1/cis2: box of height 1 μm tapering from 1 μm2 cross-section to 10 nm2; (4) DNP: length 10–20 nm, diameter 3 nm; (5) trans2: box of height 1 μm, side 1 μm. Exopeptidase covalently attached to downstream side of UNP. Electrodes at top of cis1 and bottom of trans2. V05 ≈ 0.4 V. | ||
The behavior of the proposed structure can be described qualitatively thus. A peptide with a poly-X, where X = negatively or positively charged amino acid (for pH equal to the physiological value of 7, this could be X− = Glu or Asp; X+ = Lys or Arg), leader and trailer to induce entry into UNP (V5 > V0 or V5 < V0 respectively), is drawn into UNP by a sufficiently large |V05| (about 200–400 mV typically; see Fig. 7 in ref. 1), and translocates through UNP to encounter the exopeptidase. If the latter is an amino exopeptidase the leading residues at the N-terminal of the chain are cleaved one after the other. With a carboxy exopeptidase the cleaving is at the C-terminal. (The incorrect end could enter UNP, see ESI† for how this may be resolved.) A non-zero potential difference between trans1/cis2 and trans2 causes ionic current to flow through DNP. A cleaved residue passes through DNP under the influence of V34 and/or diffusion, causing a blockade of the ionic current which can be measured. Additionally, the mean inter-arrival time (≈E(Ttrans1/cis2)) between successive cleaved residues arriving at DNP, and the mean residence time of the cleaved residue inside DNP (≈E(TDNP)) (or their variants) can also be measured.
Fokker–Planck model of the tandem cell
The mathematical model for the tandem cell here is similar to that for the tandem cell proposed earlier for exosequencing of DNA.9 The details may be found in the ESI;† only the essential features are presented here. A cleaved residue is considered to be a particle that does not interact chemically with the pore lumen or the electrolyte and moves through a combination of diffusion and electric drift. It cannot regress into UNP because it is blocked by the remaining peptide in UNP. With most of the potential difference V05 dropping across the two pores (V05 = 0.365 V, V23 = 1.6 mV, V34 = ∼0.18 V), movement through trans1/cis2, DNP, and trans2 is dominated by diffusion and can be studied via the trajectory of a particle whose propagator function G(x,y,z,t) is given by a linear Fokker–Planck (F–P) equation in one dimension (z) for DNP, or three (x,y,z) for trans1/cis2, with a drift term in the z direction that arises from V05. A piecewise approach is taken, with each section considered independent of the others. The behavior at the interface between two sections is examined later and in the ESI.† The equation is solved independently in different sections and analyzed using standard methods from partial differential equations and Laplace transforms. Translocation statistics are derived in a straightforward way.Multiple discriminators in sequencing
The ability of the tandem cell to correctly identify residues cleaved from a peptide depends on the tandem cell being able to discriminate among 20 types of residues. This may be possible if multiple discriminators in the recorded signal are used. Thus going beyond the current blockade, analyte-specific information may also be found in the times taken for a molecule or cleaved monomer to travel to a pore and through the pore. In a tandem cell both these times (or their variants) are clearly defined (translocation from top center of trans1/cis2 to DNP and translocation through DNP) and can be measured. Thus three discriminators, namely the mean blockade current ratio 〈I/I0〉 (where I and I0 are the currents with and without analyte in the pore), the mean translocation time E(Ttrans1/cis2), and the mean residence time E(TDNP) in DNP, can in principle be used in combination for analyte identification in a tandem cell. Their characteristics are discussed next.Level of current blockade inside DNP and volume excluded
Current blockade is defined by the mean blockade current ratio 〈I/I0〉. For polymer sequencing based on current blockades to work there must be an ionic current (due to K+ and Cl−) between trans1/cis2 and trans2; thus V34 cannot be 0. The blockade level is influenced by many factors, one of which is volume exclusion, whereby the particle reduces the pore volume available for ionic current flow. The volume exclusion ratio is defined as Vexcl/Vpore. Although it ordinarily contributes only a small fraction to 〈I/I0〉, here it is used as a placeholder and included in the discussion below for the purpose of studying the efficacy of multiple discriminators. When experimentally obtained or theoretically calculated values for 〈I/I0〉 become available, it may be replaced with 〈I/I0〉, after which the model can be revised as appropriate. The VER is given by eqn (5) in the ESI.†Translocation time through DNP
This is a function of the diffusion constant Daa for an amino acid, its hydrodynamic radius Raa, and mobility μaa. Daa and μaa are calculated as described in the ESI (eqn (8)†), with values Raa taken from a report on diffusion constants of the amino acids;17 the results are shown in Tables 1 and 2 in the ESI.† It is also influenced by the selectivity of the pore for anions or cations (see discussion below). Additionally the translocation can be slowed down if a chemical adapter is used.18 The mean and standard deviation of this translocation time are given by eqn (1) through 4 in the ESI.†Translocation time through trans1/cis2
The behavior of this time and its dependence on physical–chemical properties are similar to (b).Computed results
Calculated data for discriminators are given in Table 4 in the ESI† for three pH values: 7 (physiological), 5 (toward acidic), and 9 (toward basic). Fig. 2, 3, and 4 show E(TDNP), E(Ttrans1/cis2), and Vexcl/VDNP for the 20 amino acids. In the first two cases the amino acids are seen to fall into three groups, and within each group they are monotonically ordered and separate into well-defined groups. This ordering property can be used to identify residues in a peptide. (Note the behavior of cysteine, tyrosine, and histidine when pH is equal to 9.0.) The linear ordering is especially useful because error correction merely requires an incorrect call to be replaced with the nearest neighbor in the ordering. Furthermore, if the voltage V05 is reversed the two end groups reverse position in Fig. 2; this property can be used to advantage in sequencing as discussed below. (A comparison with the amino acid separation spectrum obtained from ion mobility spectrometry,19 which shows a strict mobility-based ordering of the amino acids over drift time with values in the millisecond range, reveals similar tendencies between the two orderings and some overlapping segments.)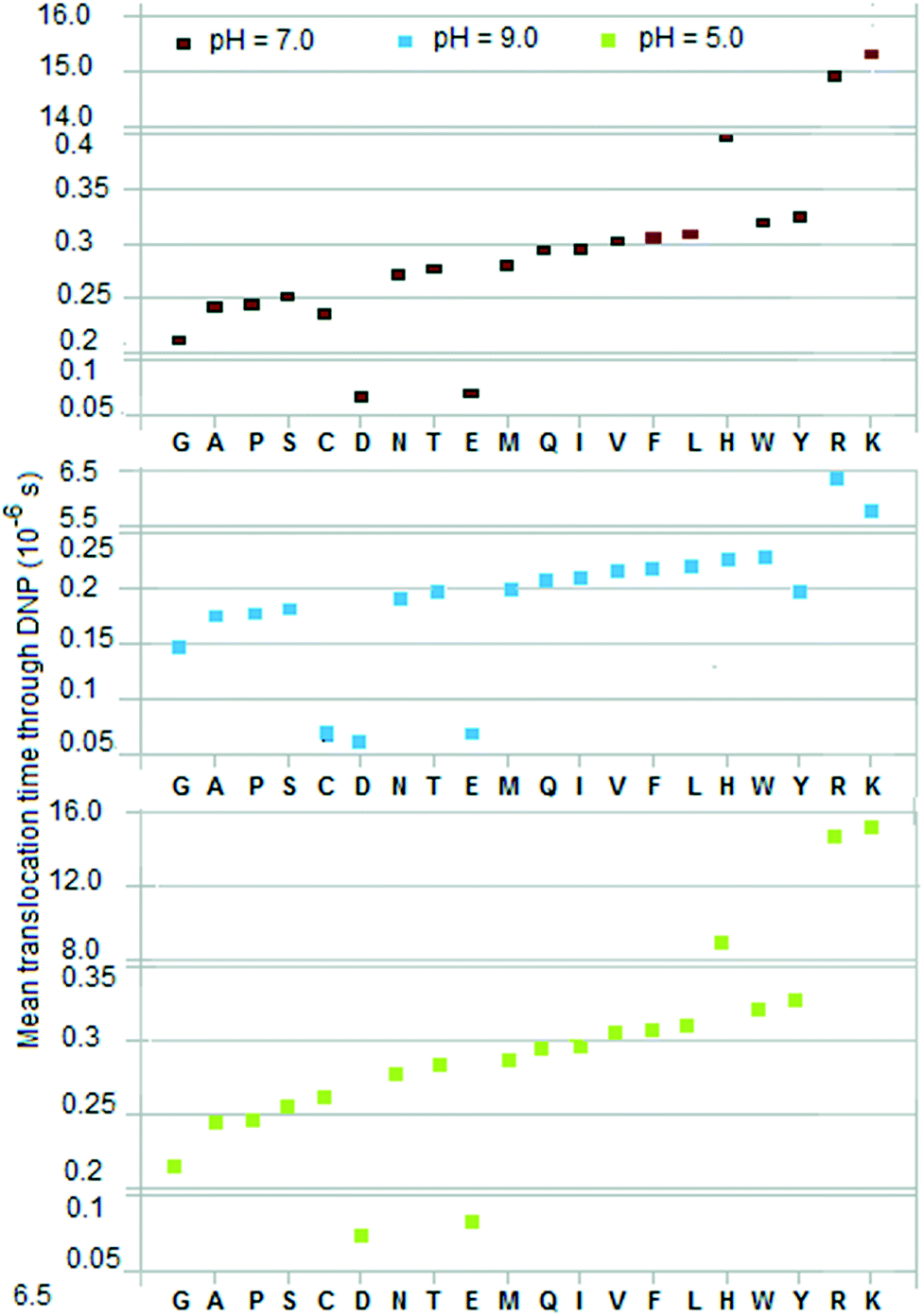 | ||
| Fig. 2 Mean of translocation time of residue from time of entry into DNP (length L34 = 10 nm, negligible cross-section) to time of exit into trans2. V05 = 0.365 V, V34 = ∼0.18 V. | ||
The discriminators described above are computed measures. The experimentally measurable quantities are somewhat different.
(1) Rather than Ttrans1/cis2 what is measured is the inter-arrival time between successive residues arriving at the pore. This quantity is Ttrans1/cis2 + Tgen, where Tgen is the time for a residue to be generated at the top of trans1/cis2 and is, like Ttrans1/cis2, a random variable. (In the tandem cell Tgen is to be replaced with the time Tc to cleave a residue from a peptide; see below.) The inter-arrival time thus contains more information than Ttrans1/cis2 because generation/cleaving times may vary with the amino acid;
(2) The time spent by a residue inside DNP is more than the translocation time TDNP because of the additional dwell arising from the reaction of the residue with the pore wall (which in a biological pore is a protein that may contain charged residues). Additional dwell time may occur if a chemical adapter (similar to cyclodextrin in DNA sequencing18) is used to slow down the residue;
(3) Current blockade, which reflects the change in the amplitude of the pore current from the baseline value, is, as mentioned above, determined by many more factors than volume exclusion. An important one is the presence of charged residues in the pore lumen (often by design20). Thus positively charged residues in the lumen will slow down negatively charged cleaved residues and vice versa, but cleaved residues with charge ≈ 0 are not affected either way.
Wet experiments may be done with free amino acids in a tandem cell or single electrolytic cell to confirm (or not) the ordering and grouping in Fig. 2 and 3.
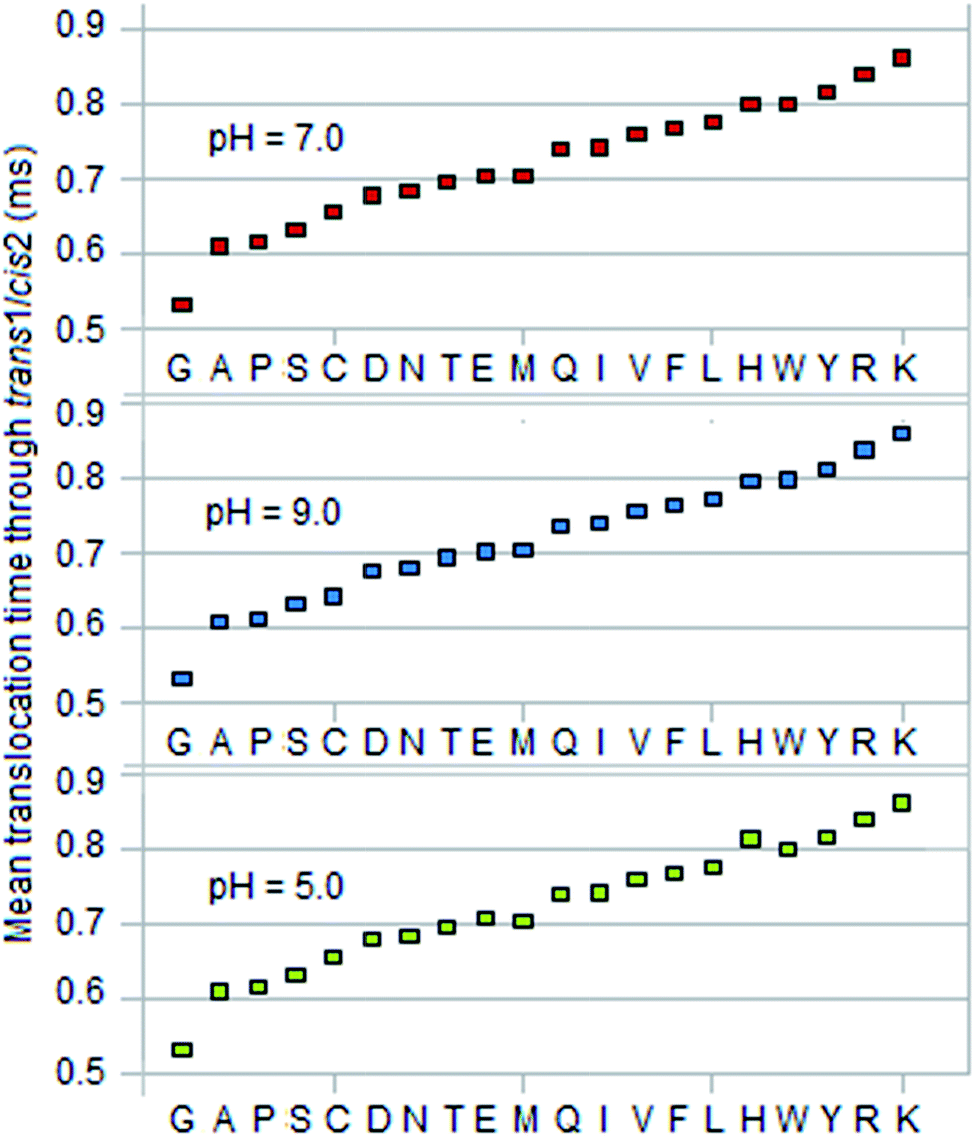 | ||
| Fig. 3 Mean of translocation time of residue to translocate from top of trans1/cis2 (length L23 = 1 μm, cross-section area = 1 μm2) to entrance of DNP. V05 = 0.365 V, V23 = ∼1.6 mV, V34 = ∼0.18 V. | ||
Behavior at an interface
The F–P model outlined above does not consider the behavior of the particle at the interface between two sections. In reality a particle oscillates at an interface because of diffusion. A residue with a substantial negative charge will eventually pass into DNP, such passage being aided indirectly by the reflecting boundaries in trans1/cis2. The behavior at the interface between DNP and trans2 is similar. Residues that are substantially positive experience a negative drift field inside both regions. There is thus a non-zero probability that such a residue may ultimately not enter DNP and therefore may be ‘lost’ to diffusion in trans1/cis2. One possible solution is to redo the sequencing using a second copy of the peptide with the voltage reversed (if the pore is ion-sensitive, one with the appropriate sense is to be used). In this case the roles of positive and negative residues are reversed. Thus the more positive residues are ‘lost’ to diffusion when V05 > 0 while the more negative residues are ‘lost’ to diffusion when V05 < 0. Thus two sequences are obtained, with some positive residues missing in one and some negative residues missing in the other, while residues with net charge ≈ 0 will appear in both sequences slightly offset from each other. The correct sequence can be obtained by merging the two sequences. See the ESI† for additional notes on this aspect of the model.Mean translocation times via sampling
The two time-based discriminators discussed above are mean values, which are to be obtained by sampling, i.e., by sequencing a peptide several times. (See ESI† for a discussion of the underlying statistics and the formulas used.) A mean value that is used to identify a residue X has to be distinguished from that of its nearest neighbor Z (the amino acid whose mean is closest to the mean of X) within a margin of error e. (This nearest neighbor can in almost all cases be identified visually in Fig. 2 and 3, where the amino acids separate into ordered groups.) Tables 5 and 6 in the ESI† give the required sample sizes for DNP and trans1/cis2 for each amino acid X and its nearest neighbor Z for three confidence levels: 90%, 80%, 70%. σ is taken from Table 4,† e = 0.4 × min|EX − EZ| where Z is the amino acid for which the mean EZ in, for example, column 2 or 4 for pH = 7.0 is closest to the mean EX for X.Fig. 4, 5 and 6 show histograms of the sample size for DNP and trans1/cis2 respectively. The value of N to use in the sequencing is the largest sample size Nmax over all the amino acids. Amino acid pairs whose mean times are very close to each other are the ones that effectively determine Nmax. As seen from Tables 5 and 6† (or Fig. 2 and 3) the problem pairs in the case of DNP are His (H)–Trp (W), Gln (Q)–Ile (I), Met (M)–Tyr (Y), and Ala (A)–Pro (P), with N in the range 1 to 6 million; in the case of trans1/cis2 they are Glu (E)–Met (M), His (H)–Trp (W), and Gln (Q)–Ile (I) with N in the range 1 to 11 million. A more manageable value of Nmax is possible if these highly error-prone residue pairs are excluded from its determination. This lowers the confidence levels for their measured means but their identification can be handled through error correction (made easier by the ordering property; see Fig. 2 and 3). Such error correction could be based on, for example, methods used in mass spectrometry.10 In the case of DNP this leads to Nmax = ∼70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for a confidence level of 90% or better for the other 12 amino acids, and ∼27000 for a confidence level of 70% or better. For a long peptide, Nmax could in principle be lowered by a factor of Lpep/20, where Lpep is the peptide length, because of repeats; this assumes that the 20 amino acids occur in proteins with equal probability.
000 for a confidence level of 90% or better for the other 12 amino acids, and ∼27000 for a confidence level of 70% or better. For a long peptide, Nmax could in principle be lowered by a factor of Lpep/20, where Lpep is the peptide length, because of repeats; this assumes that the 20 amino acids occur in proteins with equal probability.
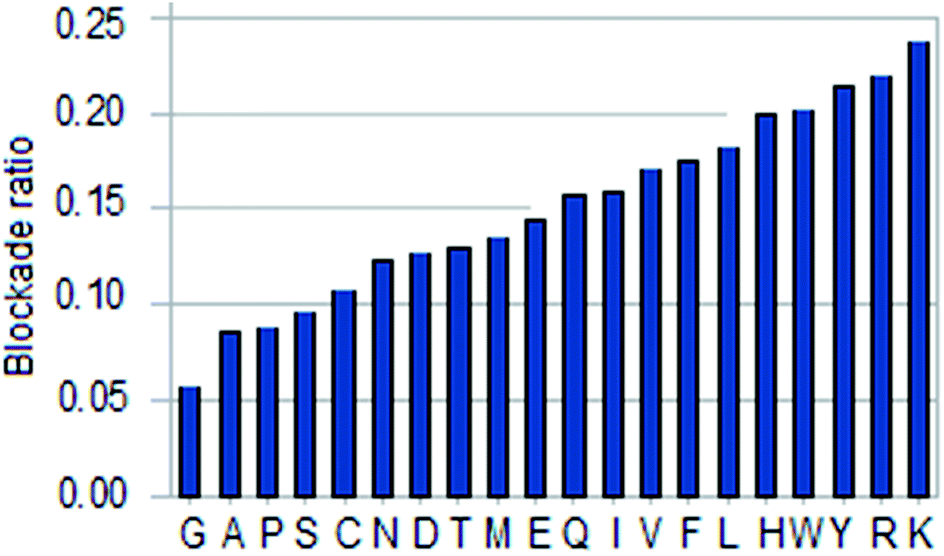 | ||
| Fig. 4 Histogram of amino acid blockade ratio. | ||
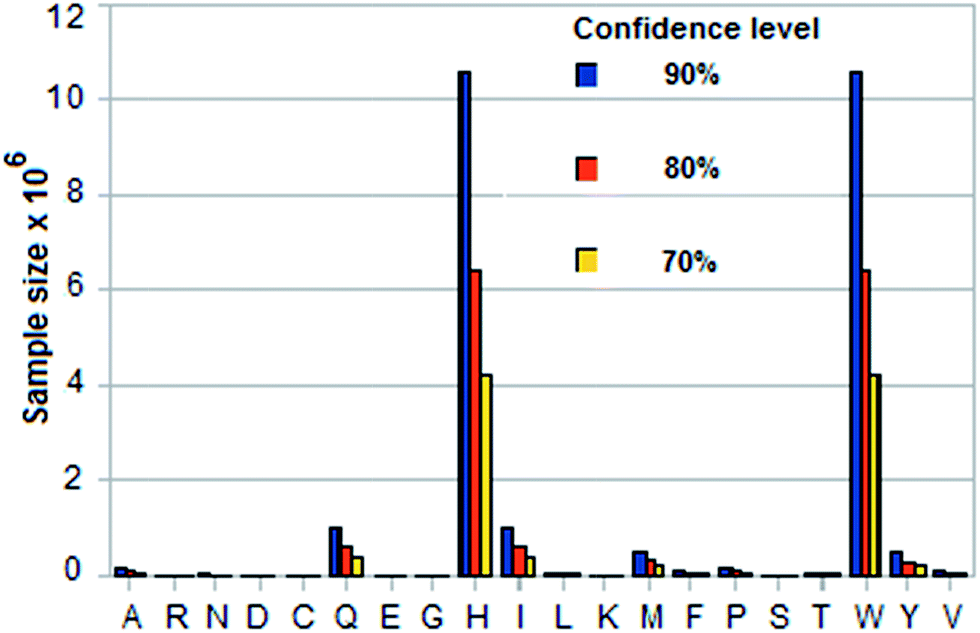 | ||
| Fig. 5 Sample sizes for three confidence levels for each amino acid based on standard statistical formula involving standard deviation of its translocation time in DNP and margin of error = 0.4 × smallest difference between its mean translocation time and that of any of the other 19. See Tables 4 and 5 in ESI† for calculated data (pH = 9.0). | ||
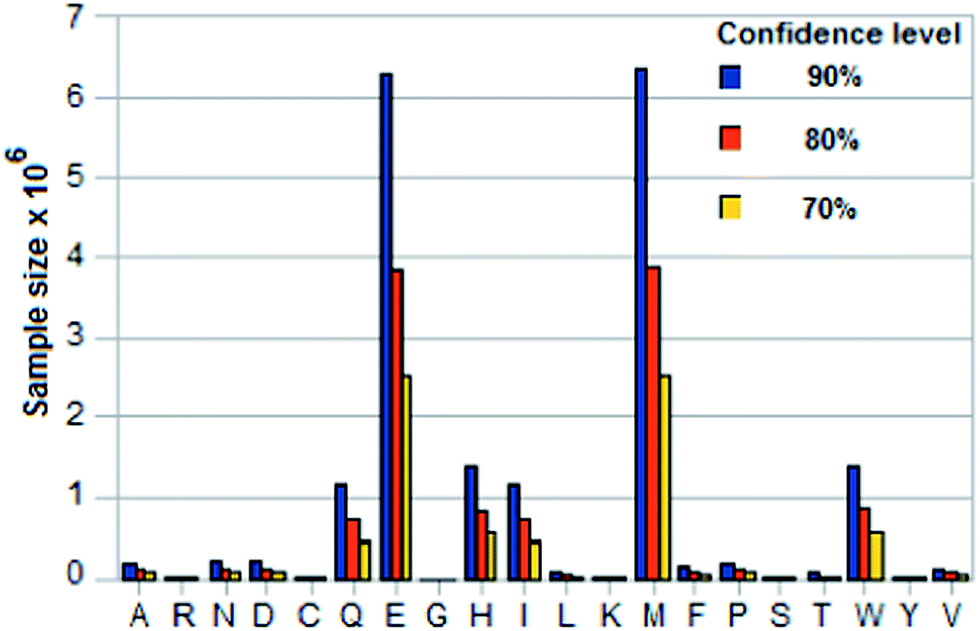 | ||
| Fig. 6 Sample sizes for three confidence levels for an amino acid based on standard statistical formula involving standard deviation of its translocation time in trans1/cis2 and margin of error = 0.4 × smallest difference between its mean translocation time and that of any of the other 19. See Tables 4 and 6 in ESI† for calculated data (pH = 9.0). | ||
By way of comparison, the reduced value of ∼70000 for Nmax is of the same order as the number of crystals used in serial femtosecond nanocrystallography21 (SFX) to determine protein structure using a ‘diffract-then-destroy’ approach. SFX uses a high intensity laser pulse to capture the diffraction image of one of ∼104 crystals in a liquid jet (LJ-SFX) injected by a nozzle into the path of the laser or on a fixed target (FT-SJX) interposed mechanically. The entire sample is destroyed in the process, but not before the image is captured.
Conversely for a given maximum number of samples Nmax one can find the confidence level for the sample mean of an amino acid X to be no farther from the population mean than k × e, where e is the distance to the nearest mean, with k < 0.5. Table 7 in the ESI† gives the confidence levels for the 20 amino acids for k = 0.4 and N = 10000 in DNP and trans1/cis2. Fig. 7 shows a histogram of comparative confidence levels of residue identification in DNP and trans1/cis2 for all 20 amino acids for pH = 9.0.
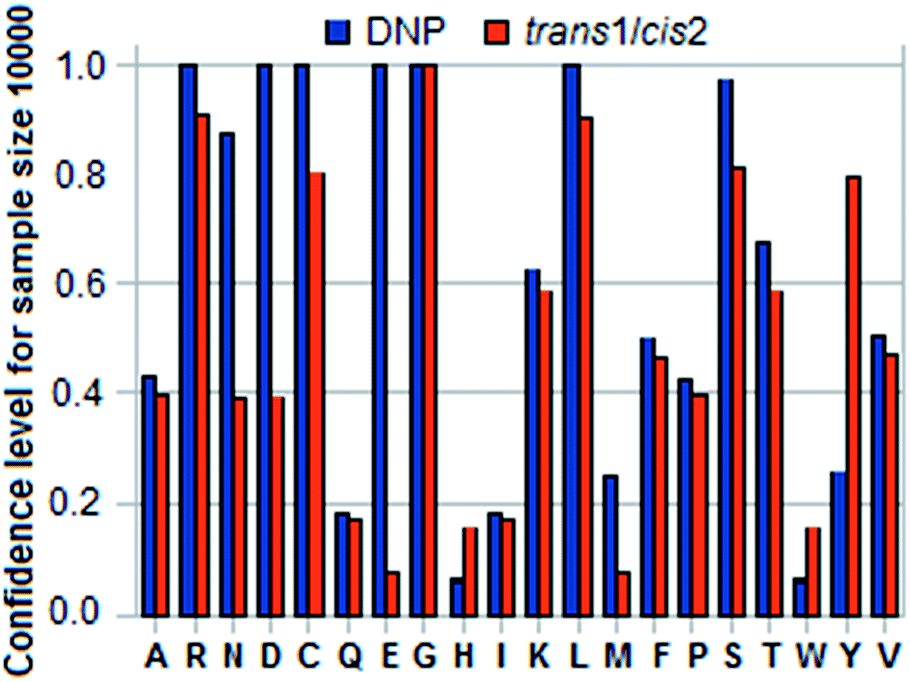 | ||
| Fig. 7 Histogram of confidence levels for an amino acid X to ensure that sample mean translocation time in DNP or trans1/cis2 is within 0.4 × smallest difference between the calculated mean for X and that for any of the other 19 for a sample size of 10000 and pH = 9.0. See Table 7 in ESI.† | ||
Assuming ergodicity and the availability of a sufficient quantity of the assay sample a parallel implementation of Nmax tandem cells can be used with Nmax copies of the peptide to quickly obtain the sample mean for every residue in the peptide. Such an approach (which would need to be automated because of the large values of Nmax involved) would be more appropriate to research than to clinical or forensic assays where only a limited amount of the test sample may be available. Alternatively, with only one sample the sequence of cleaved residues entering trans2 may be recycled to cis1 for re-sequencing. This possibility is discussed in the ESI.†
Necessary conditions for accurate sequencing
For accurate sequencing the following conditions must be satisfied: (1) residues must not be lost to diffusion; (2) residues must enter DNP in sequence order; (3) no more than one residue may occupy DNP at any time. It is shown in the ESI† (Section 5) that these conditions are met for the parameter values assumed. Conditions 2 and 3 also serve to define the minimum interval required between successively cleaved residues in the peptide:| Tc min ≈ 2.99 ms | (1) |
Properties of exopeptidase
The feasibility of the proposed scheme depends on the exopeptidase being able to cleave every (leading) residue in the peptide. Serine carboxypeptidases appear to have this kind of capability, in particular carboxypeptidase Y (CPD-Y) and malt carboxypeptidase II (CPD-M-II); see review.22 Similarly leucine aminopeptidases (LAP) have the ability to cleave a range of amino acid types.23 CPD-Y and CPD-M-II have been reported as cleaving leading residues successively at the C-terminal fairly reliably, although the specificity (or equivalently the cleavage intervals) varies widely and depends on both the pH (typically 5.0 to 7.0) and the substrate (that is, the peptide to be sequenced). Thus CPD-Y has a preference for hydrophobic residues and CPD-M-II for basic; both are slow in releasing Thr, Ser, Asn, Gln, Glu, Asp, and Pro.24 Note the large sequencing times reported, typically several minutes, which is in part because sequencing is in the bulk and involves two disjoint steps: residue cleaving and spectrophotometry assay.24 Also if residues are cleaved too fast, sequence information gets scrambled. Neither of these problems would occur in a tandem cell because: (a) nanopore sequencing is a single molecule method in which an individual polymer strand is sequenced or identified using only electrical measurements in an integrated process; and (b) the tandem cell has a pipeline structure in which cleaved residues are separated in time and space and retain their natural order as long as the ‘Necessary conditions’ mentioned above are satisfied.The literature on exopeptidases is large and scattered; exopeptidases other than those considered here may be located by a search of the biochemical literature. It remains to be seen if the exopeptidases described above can be used with similar success in a tandem cell, which requires the enzyme to be covalently attached to the downstream side of UNP. One could also consider attaching two peptidases with different specificities (for example, CPD-Y and CPD-M-II) to UNP, although doing this in a reliable way might be tricky. A better and perhaps easier alternative would be sequencing two copies in two different tandem cells each with a different exopeptidase and merging the resulting sequences. As suggested in the review mentioned above,22 it may be possible to engineer the enzymes themselves to achieve the intended result. For example, changing a few residues might lead to significant improvement in performance and this may be done through trial and error. Hopefully a clearer picture may emerge from experiments with a tandem cell involving a wide range of parameters (pH, applied voltage, temperature, etc.).
Peptides with modified amino acids
While most studies of peptide sequencing are concerned with identification of the 20 naturally occurring amino acids, in practice one has to consider the wide variety of modified amino acids that occur in nature, in particular those that arise from post-translational modifications in the cell. Modified amino acids can be taken into account in the tandem cell approach presented here without any major modifications. The most significant change to expect would be a decrease in discrimination capability, a natural consequence of an increase in the number of monomer types. This translates to a decrease in the minimum mean distance between two residues and a consequential increase in the sample size required.Implementation issues
The following factors may be considered in a practical implementation. See ESI† for an expanded discussion.(1) With translocation times through DNP on the order of 10−7 s (see Table 4†), the bandwidth required is ∼10 MHz (including noise filtering). The lower signal-to-noise ratio in this frequency range combined with the pA-sized blockade levels and fast translocation makes detection difficult (this problem exists in nanopore-based sequencing of any analyte, including DNA). Methods to slow down translocation in a tandem cell have been discussed elsewhere.9 As noted earlier, charged residues in the pore lumen (by design) can be used to slow down a translocating charged analyte.20
(2) As mentioned earlier, Hidden Markov Models, Viterbi algorithms, and complex neural nets12,13 have been used to increase base calling accuracy in strand sequencing of DNA. They can be modified to work with the multiple discriminators discussed above for improving residue calling accuracy.
(3) The proposed scheme assumes that with amino (carboxy) exopeptidase the peptide enters UNP N-terminal (C-terminal) first. There is no guarantee that this will happen. See ESI† for how this problem could be resolved.
(4) The optimum peptide length handled by an efficient mass spectrometer10 is ∼20. Considerably longer reads may be possible with a tandem cell.
(5) It is possible for some neutral residues to attract ions in an electrolyte and carry a resulting charge.14,25 A cleaved residue that is ordinarily neutral can therefore become positively or negatively charged due to formation of an anion or cation complex. No information is available about whether amino acids form such complexes in aqueous KCl or not, so this line of investigation has not been pursued.
(6) A folded protein loaded into cis1 could be unfolded by an unfoldase enzyme5 like ClpX before it is cleaved and sequenced. This raises the possibility of whole protein sequencing in a tandem cell.
(7) Transverse recognition tunneling8 (RT) may be adapted for use with a tandem cell. Thus the leading residue cleaved by the exopeptidase attached to UNP could be identified by the transverse current through a recognition molecule in DNP without being disadvantaged by low ionic current blockade levels, high bandwidth requirement because of fast translocation, and low signal-to-noise ratios.1
(8) For other implementation-related issues in sequencing with a tandem cell, such as voltage drift and bases/monomers that could stick to channel walls, and their possible resolution, see discussion elsewhere.9
Acknowledgements
The author thanks the referees for their helpful comments and suggestions.References
- M. Wanunu, Physics of Life Reviews, 2012, 9, 125–158 CrossRef PubMed.
- W. Timp, A. M. Nice, E. M. Nelson, V. Kurz, K. Mckelvey and G. Timp, IEEE Access, 2014, 2, 1396–1408 CrossRef.
- J. Nivala, D. B. Marks and M. Akeson, Nat. Biotechnol., 2013, 31, 247–250 CrossRef CAS PubMed.
- A. Oukhaled, L. Bacri, M. Pastoriza-Gallego, J.-M. Betton and J. Pelta, ACS Chem. Biol., 2012, 7, 1935–1949 CrossRef CAS PubMed.
- J. Nivala, L. Mulroney, G. Li, J. Schreiber and M. Akeson, ACS Nano, 2014, 8, 12365–12375 CrossRef CAS PubMed.
- C. B. Rosen, D. Rodriguez-Larrea and H. Bayley, Nat. Biotechnol., 2014, 32, 179–181 CrossRef CAS PubMed.
- D. Wu, S. Bi, L. Zhang and J. Yang, Sensors, 2014, 14, 18211–18222 CrossRef CAS PubMed.
- Y. Zhao, B. Ashcroft, P. Zhang, H. Liu, S. Sen, W. Song, J. Im, B. Gyarfas, S. Manna, S. Biswas, C. Borges and S. Lindsay, Nat. Nanotechnol., 2014, 9, 466–473 CrossRef CAS PubMed.
- G. Sampath, RSC Adv., 2015, 5, 167–171 RSC.
- H. Steen and M. Mann, Nat. Rev., 2004, 5, 699–711 CrossRef CAS PubMed.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W H Freeman, New York, NY, 5th edn, 2002 Search PubMed.
- W. Timp, J. Comer and A. Aksimentiev, Biophys. J., 2012, 102, L37–L39 CrossRef CAS PubMed.
- A. H. Laszlo, I. M. Derrington, B. C. Ross, H. Brinkerhoff, A. Adey, I. C. Nova, J. M. Craig, K. W. Langford, J. M. Samson, R. Daza, K. Doering, J. Shendure and J. H. Gundlach, Nat. Biotechnol., 2014, 32, 829–834 CrossRef CAS PubMed.
- B. Lu, D. P. Hoogerheide, Q. Zhao, H. Zhang, Z. Tang, D. Yu and J. A. Golovchenko, Nano Lett., 2013, 13, 3048–3052 CrossRef CAS PubMed.
- M. Schiel and Z. S. Siwy, J. Phys. Chem. C, 2014, 118, 19214–19223 CAS.
- J. Clarke, H.-C. Wu, L. Jayasinghe, A. Patel, S. Reid and H. Bayley, Nat. Nanotechnol., 2009, 4, 265–270 CrossRef CAS PubMed.
- M. W. Germann, T. Turner and S. A. Allison, J. Phys. Chem. A, 2007, 111, 1452–1455 CrossRef CAS PubMed.
- A. Banerjee, E. Mikhailova, S. Cheley, L.-Q. Gu, M. Montoya, Y. Nagaoka, E. Gouauxd and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8165–8170 CrossRef CAS PubMed.
- G. R. Asbury and H. H. Hill Jr, J. Chromatogr. A, 2000, 902, 433–437 CrossRef CAS.
- T. Z. Butler, M. Pavlenok, I. M. Derrington, M. Niederweis and J. H. Gundlach, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20647–20652 CrossRef CAS PubMed.
- M. S. Hunter, B. Segelke, M. Messerschmidt, G. J. Williams, N. A. Zatsepin, A. Barty, W. H. Benner, D. B. Carlson, M. Coleman, A. Graf, S. P. Hau-Riege, T. Pardini, M. Seibert, J. Evans, S. Boutet and M. Frank, Sci. Rep., 2014, 4, 6026, DOI:10.1038/srep06026.
- K. Breddam, Carlsberg Res. Commun., 1986, 51, 83–128 CrossRef CAS.
- A. Taylor, FASEB J., 1993, 7, 290–298 CAS.
- K. Breddam and M. Ottesen, Carlsberg Res. Commun., 1987, 52, 55–63 CrossRef CAS.
- J. E. Reiner, J. J. Kasianowicz, B. J. Nablo and J. W. F. Robertson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12080–12085 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Mathematical details and calculations, an expanded discussion, and tables of calculated data (including data for nucleotides for comparison). See DOI: 10.1039/c5ra02118a |
| This journal is © The Royal Society of Chemistry 2015 |