
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Platinum nanoparticles supported on N-doped carbon nanotubes for the selective oxidation of glycerol to glyceric acid in a base-free aqueous solution†
Shasha
Chen
,
Puyu
Qi
,
Jin
Chen
and
Youzhu
Yuan
*
State Key Laboratory of Physical Chemistry of Solid Surfaces and National Engineering Laboratory for Green Chemical Productions of Alcohols-Ethers-Esters, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: yzyuan@xmu.edu.cn
First published on 27th March 2015
Abstract
Selective oxidation of glycerol was carried out over Pt catalysts supported on nitrogen-doped carbon nanotubes (Pt/N-CNTs) with molecular oxygen under atmospheric pressure in base-free aqueous solution. The N-CNTs were readily synthesized through a catalyst-free approach of annealing the mixture of CNTs and melamine. Results of X-ray diffraction, nitrogen absorption, and Raman spectroscopy confirmed that the tubular structure of CNTs was intact during annealing. Analyses of transmission electron microscopy, X-ray photon spectroscopy, and temperature-programmed desorption indicated that the surface of the CNTs was successfully functionalized with nitrogen atoms, which changed the electronic structure and surface basicity of the N-CNTs. Pt/N-CNTs out-performed Pt/CNTs for glycerol oxidation in terms of glycerol conversion and glyceric acid selectivity. Pt/N-CNTs showed highly stable catalytic performance during consecutive recycles when the used catalyst was reduced in a H2 atmosphere.
1. Introduction
With the depletion of fossil fuels and gradual improvement in environmental protection consciousness, biodiesel fuels have been receiving worldwide attention and are being rapidly developed for their superior environmental performance, renewability, and nontoxic advantages.1,2 Glycerol is a by-product in the manufacture of biodiesel through the transesterification of fatty acids. Every ton of biodiesel production is known to produce 100 kilograms of glycerol.3,4 Thus, the rapid development and commercialization of biodiesel may cause a surplus of glycerol. To this end, catalytic conversion of glycerol into value-added products has attracted considerable attention from the aspects of environmental protection and economic benefit. In the past years, numerous reports focused on the selective transformation of glycerol to various useful chemicals via oxidation, reduction, halogenation hydrogenolysis, dehydration, pyrolysis, transesterification and so on.5 Among them, numerous studies have been conducted for the chemoselective oxidation of glycerol with molecular oxygen to ponderable products such as glyceric acid (GLYA), glyceraldehyde (GLYDE) and dihydroxyacetone (DHA).6Noble metal particles, such as Pt, Pd, and Au, loaded on carbon materials like activated carbon and carbon nanotubes (CNTs) are the well-known efficient catalysts for glycerol oxidation under pressurized oxygen and/or alkaline conditions. Studies have shown that catalytic performance definitely depends on the type of noble metal, but more strongly on the basicity of the reaction medium. In certain cases, however, the reaction produced large amounts of undesired C1 by-products, such as CO2, HCHO, and HCOOH, in alkali solution.7–11 For instance, Gallezot et al. reported that selectivity toward GLYA reached 70% on Pd/C at the optimized pH of 11, whereas this value decreased to 55% on Pt/C at the optimized pH of 7.12 Au-based catalysts were also investigated under strong basic conditions (NaOH/glycerol >1),9,13–16 Prati et al. revealed that even well-dispersed Au nanoparticles on carbon with a mean diameter of 6 nm cannot maintain its initial selectivity at full conversion, and that Au is totally inactive without the help of NaOH.17,18
The addition of a base, such as NaOH, contributes to the capture of H+ from primary hydroxyl groups of glycerol to initiate reaction.19–21 Meanwhile, bases promote the adsorption and activation of glycerol on the surface of Pt atoms.20 Moreover, during reaction, the irreversible adsorption of the reaction products on the catalyst surface is inevitable, and the presence of a base can remove such products from the active surface, thus effectively prolonging catalyst life.22,23 However, glycerate, instead of GLYA, is formed when a homogeneous base is used, and the reaction liquid requires an additional neutralization or acidification step prior to analysis in a high-performance liquid chromatograph (HPLC). The use of a base complicates the operation process and may lead to environmental pollution. Furthermore, peroxide, which is related to the cleavage of C–C bonds and the formation of C1 by-products, is also formed in basic conditions, and its amount depends on the concentration of the added base.15,24,25 Thereby, a homogeneous base should be replaced or severely avoided from the viewpoint of environmental protection and green chemistry.
To avoid the addition of bases, several researchers adopted basic materials like hydrotalcite, MgO, MgAl2O4, and Al2O3 to replace the carbonaceous supports, thus permitting base-free oxidation and producing carboxylic acid, rather than the salt form.26–29 However, the poor resistivity of these materials in acid solution causes leaching of supported active metal species, resulting in limited stability and lifetime of the corresponding catalysts.
CNTs have been extensively studied as a carrier for metal nanoparticles in the catalytic oxidation of glycerol because of its excellent electronic properties, good physical and chemical stability, and large surface area.15,30 However, given their surface inertness and their insufficient anchoring sites for binding metal nanoparticles, CNTs usually requires a suitable surface functionalization pretreatment for better immobilization of catalytic particles. In recent years, nitrogen-containing carbon nanotubes (N-CNTs) have attracted considerable attention for their outstanding physicochemical properties, such as the following: (1) provides versatile reactive sites for a robust interface; (2) lowers the work-function for facile charge injection and redox reaction; (3) modifies the charge transfer characteristics with excessive electrons; (4) offers permanent dipoles that can activate catalytic activity and surface reactivity; and (5) can alter the surface acid/base properties of CNTs by functionalization via amination.31 N-atom can modify the electronic structure of CNTs with delocalization effects at the N-site, thus inducing localized charge accumulation. Localized charge density functions in the electron transfer reaction and facilitates the adsorption and/or dissociation of molecules, and imparts N-CNTs with a particular importance in catalysis.32–35 In the literatures,33,36 N-CNTs performed well in oxygen reduction reactions and electrochemical catalysis. However, the hydrophobicity of CNTs may be decreased after the incorporation of the N-atom.36 Regardless, the incorporation of N in the graphitic structure of carbon materials represents a means of inducing basic surface properties in heterogeneous catalysis,32 which may be beneficial for the oxidation of glycerol in base-free aqueous solutions.
Herein, we report the catalytic performance of Pt nanoparticles loaded on N-CNTs for the liquid-phase oxidation of glycerol with molecular oxygen under atmospheric pressure and in base-free conditions. The surface properties of CNTs after doping N-atom and their impacts on catalytic activity are addressed in detail.
2. Experimental
Catalyst preparation
CNTs (20 nm to 40 nm in diameter) with a purity of 95% were purchased from Shenzhen Nanotechnologies Port Co. Melamine was supplied from Alfa Aesar Ltd. C3N4 was synthesized by the calcination of melamine under air in a muffle furnace.37 Chloroplatinic acid (H2PtCl6·6H2O), was purchased from Sinopharm Chemical Reagent Co., Ltd. GLYA, GLYDE, and DHA were purchased from J&K Technology Co., Ltd.Pristine CNTs were purified and functionalized in concentrated HNO3 (68 wt%) at 80 °C for 16 h under refluxing conditions to remove amorphous carbon and the remaining catalyst residues. The treated CNTs were filtered, extensively washed with deionized water until the pH of the rinsing water became neutral, and then dried at 110 °C overnight. These CNTs were designated as CNTs-353.
N-CNTs were commonly synthesized by catalytic chemical vapor deposition (CCVD) method on the supported transition-metal based catalysts.32,38,39 However, several difficulties existed during the process of controlling growth, which invariably led to a mixture of tubes and bamboo-like fibers. On the other hand, the transition-metal based catalyst would contaminate the resultant products and thus affect their properties. Another synthesis route involves the amination of CNTs with long-chain organic amines, NH3 or HCN.22,39 Besides improving the solubility and dispersion of CNTs, the method could also introduce the desired basic moieties. However, NH3 and HCN are toxic, and should be avoided for safety. Herein, we selected a facile and catalyst-free approach of annealing the mixture of CNTs and melamine to synthesize N-CNTs.41 Compared with the CCVD method, this post-functionalizational method can completely prevent the contamination of transition metal catalysts and presents distinct advantages. CNTs-353 and melamine are typically mixed, forming a uniform mixture after being grinded in a mortar. Wrapped in aluminum foil, the mixture was then placed in a corundum tube with a nitrogen atmosphere flow and heated to 600 °C at a rate of 2 °C min−1. After the temperature was maintained for 1 h, the furnace was gradually cooled to room temperature. The support was eventually obtained and designated as N-CNTs(x), where x represents the N content in the corresponding support as determined by elemental analysis. N-CNTs with different nitrogen concentration can be achieved by controlling the mass ratio of CNTs-353 and melamine.
Supported Pt catalysts were prepared by wet impregnation with aqueous solutions of chloroplatinic acid according to a procedure described in the literature.42 The aqueous solution of H2PtCl6 was added to N-CNTs under stirring. After 12 h of agitation at ambient temperature, the suspension was evaporated in a water bath at 60 °C to remove water. Then, the solids were dried at 110 °C for 12 h. Then, the required amount of formaldehyde aqueous solution was subsequently added. After adjusting the pH to above 10 with 0.1 M NaOH, the suspension was refluxed at 90 °C for 30 min and then cooled to room temperature. Finally, the black solids were collected by filtration, washing with distilled water, and drying at 110 °C overnight. The final catalyst was produced after reduction in a 5% H2–95% N2 atmosphere at 350 °C for 4 h and labelled as Pt/N-CNTs(x).
Glycerol oxidation
The oxidation of glycerol was conducted in a two-neck flask (25 mL) equipped with a gas supply system, a magnetic stirrer, a condenser, and a thermocouple. A 10 mL aqueous solution of glycerol (0.1 mol mL−1) and 0.039 g catalyst (glycerol/metal = 500 mol mol−1) were added into the reactor. Once temperature reached 60 °C, O2 (99.999%) was introduced into the reactor at 10 mL min−1via a mass flow controller at atmospheric pressure. After the reaction, the catalyst was filtered off, and the aqueous solution was analyzed using a HPLC (Shimadzu LC20A) with acetone as an internal standard. The HPLC was equipped with a Shodex sugar column (SH 1011) and a refractive index detector. All products were confirmed by comparison with standard products purchased from J&K Technology Co., Ltd. The turnover frequency (TOF) was based on the number of surface Pt metal atoms determined from the results of O2 titration, indicating the moles of glycerol converted by per Pt atom at the catalyst surface per hour (mol-glycerol mol-Ptsurf−1 h−1, for short, h−1). The glycerol conversion for the TOF calculation was 30–40% by decreasing the amount of catalyst and shortening the reaction time.Catalyst characterization
The N2 adsorption–desorption isotherm was measured at 77 K using a Micromeritics TriStar II 3020 porosimetry analyzer. Textural data were calculated from the desorption isotherms. Powder XRD patterns were obtained using a Philips PANAlytical X'pert Pro diffractometer equipped with a graphite monochromator and Cu Kα radiation (35 kV and 15 mA). The scanning angle ranged between 5° and 80°, the step size was 0.0167°, and the scanning time per step was 10 s. Transmission electron microscopy (TEM) images were obtained using a Tecnai F20 electron microscope operated at an acceleration voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) was performed using a Quantum 2000 Scanning ESCA Microprobe instrument (Physical Electronics) equipped with an Al Kα X-ray radiation source (hν = 1486.6 eV). Binding energy was calibrated on the base of C 1s (284.6 eV). Raman spectra of the as-synthesized N-CNTs were collected by RENISHAW LEIA 50 Laser Confocal Raman Microspectroscopy. Temperature-programmed desorption-mass spectrometry (TPD-MS) was performed using a Micromeritics AutoChem II 2920 instrument. A mass spectrometer (ThermoStar GSD 301 T2) was used to monitor CO and CO2 signals. Metal dispersion (D) was studied by employing the H2–O2 titration method using Micromeritics ASAP 2020 apparatus. Assuming that the absorption stoichiometry factor of Pt/H2 = 2/3, then Pt/O2 = 2. Thermogravimetric analysis (TGA) was conducted on TG_209F1 equipment from 30 to 1000 °C with a heating rate of 10 °C min−1 under nitrogen.3. Results and discussion
Characterization of support and catalyst
The texture structures of catalysts are summarized in Table 1. The calculated surface area of pretreated CNTs is 112.4 m2 g−1, which decreases from 93.6 m2 g−1 to 10.0 m2 g−1 (Table S1†) with increasing N content from 1.57 to 61.67. The decrease in surface area can be attributed to increased N deposition, resulting in pore blockage of CNTs. However, after the loading of Pt atoms, the SBET of the catalysts is higher than the those of corresponding supports, decreasing sharply (from 114.6 to 64.6) with increasing N content. Meanwhile, pore volume and pore diameter do not vary significantly, indicating that Pt atoms are loaded on the surface of supports and that the tubular structure of catalysts are all intact.| Sample | S BET/m2 g−1 | V pore a/cm3 g−1 | D pore/nm | Mean sizeb/nm | Dispersion (D/%) | ||
|---|---|---|---|---|---|---|---|
| D c (TEM) | D d (O2) | D e (H2) | |||||
| a Obtained from P/P0 = 0.99. b Mean size of Pt particle, statistical data by TEM characterization. c All Pt particles were regarded as spheres. Thus, Pt dispersion, D, was calculated as D = (6υm/αmd) × 100%, where D is the Pt dispersion; υm is the volume of a Pt atom, υm = M/ρNo, M is the molecular weight of Pt, ρ is the density of Pt, No is Avogadro's constant; αm is the area of a Pt atom on the surface, αm = 1/(1.25 × 1019), and d is the Pt particle diameter determined by TEM. Therefore, D = 1.13/d. d Calculated by H2–O2 titration. e Calculated by H2–O2 titration. | |||||||
| CNTs-353 | 112.4 | 0.29 | 10.2 | — | — | — | — |
| 1% Pt/CNTs-353 | 114.6 | 0.32 | 10.7 | 2.7 | 41.9 | 55.4 | 30.3 |
| 1% Pt/N-CNTs(1.57) | 101.0 | 0.37 | 13.7 | 2.2 | 51.4 | 55.6 | 33.7 |
| 1% Pt/N-CNTs(3.32) | 89.6 | 0.27 | 11.4 | 2.1 | 53.8 | 51.5 | 43.6 |
| 1% Pt/N-CNTs(5.74) | 86.5 | 0.29 | 11.4 | 2.3 | 49.1 | 47.6 | 34.5 |
| 1% Pt/N-CNTs(9.44) | 80.8 | 0.26 | 11.6 | 2.0 | 56.5 | 11.9 | 20.9 |
| 1% Pt/N-CNTs(22.85) | 64.6 | 0.18 | 9.7 | — | — | 13.3 | 23.6 |
From the XRD patterns (Fig. 1) of the catalysts, no typical diffraction peaks of Pt(111) can be detected at 39.8°, indicating that Pt particles are small and well-dispersed on the supports. For all catalysts, three major peaks observed at 26.0°, 42.9°, and 53.7° are assigned to the (002), (100), and (004) graphite planes respectively, which shows the well-graphitized nature of CNTs after the incorporation of N.
 | ||
| Fig. 1 XRD patterns of 1% Pt/N-CNTs(x) catalysts with different N content. | ||
In order to measure the Pt particle size, TEM characterization is performed, and the typical TEM images of Pt/CNTs-353 and Pt/N-CNTs(x) catalysts, as well as the histograms of particle size distribution are displayed in Fig. S1 (ESI†). We can see that Pt particles evenly load on the surfaces of CNTs-353 and N-CNTs with a uniform and narrow size distribution, and the calculated mean particle size is 2 to 3 nm (2.7, 2.1, 2.3, and 2.0 nm for 1% Pt/CNTs-353, 1% Pt/N-CNTs(3.32), 1% Pt/N-CNTs(5.74), and 1% Pt/N-CNTs(9.44) catalysts, respectively), which is in accordance with the XRD results. It seems that after the introduction of N atoms into CNTs, the Pt particles become smaller with uniformly dispersion. Reportedly, the presence of oxygen-containing surface groups after treatment with nitric acid leads to poorer metal dispersion.30 On the other hand, incorporation of N influences the electronic conductivity of CNTs by donating its electron to the CNTs, and then promotes the effective immobilization of Pt nanoparticles by strengthening the metal–support interactions through the large electron affinity of N. N-doping can significantly enhance the surface energy of graphitic carbons, which may reduce the energy barrier for heterogeneous nucleation.36,40 Moreover, Pt nanoparticles present higher binding energies in the N-doped graphitic plane than their in undoped counterpart. This higher energy is due to the strong interaction of their d-orbitals with the p-orbitals of N, thereby preventing Pt particles from sintering. The results that the presence of N functionalities can limit the growth of the metal nanoparticles have also been reported in literature.33,43,44 Further experiments by elemental STEM-EDX mappings (Fig. 2) of the catalysts also convince the above results. Therefore, we can conclude that N atoms are not only introduced into the CNTs successfully but are also spread in the material evenly along the distribution of C atoms. In addition, Pt particles are loaded on the surface of support uniformly, as well.
 | ||
| Fig. 2 TEM dark-field image and STEM-EDX elemental mappings of 1% Pt/N-CNTs(5.74) catalyst. (a) Dark-field image. Other images represent the elemental STEM-EDX mapping images of measured C–K intensity, N–K intensity, and Pt-L intensity, which are obtained from the light brown square. The yellow rectangle is used to correct any drift in the sample. | ||
XPS characterizations are further performed to analyze the elemental composition and N bonding configurations in N-CNTs. From the XPS profile (Fig. 3A) of CNTs-353, only the signals assigned to C and O atoms at 284.6 and 532.2 eV, respectively, can be observed. However, on the XPS profile of N-CNTs, another apparent peak appears at 401.1 eV, which belongs to N atoms, and the intensity of the peaks is enhanced with increasing N content. This phenomenon strongly suggests the presence of additional components, namely, N atoms, which agrees with TEM characterization.
 | ||
| Fig. 3 XPS profiles of N-CNTs with different N content: (A) wide scan of XPS profiles; (B) C 1s XPS profiles; (C) N 1s XPS profiles. (a) CNTs-353, (b) N-CNTs(1.57), (c) N-CNTs(3.32), (d) N-CNTs(5.74), (e) N-CNTs(9.44), and (f) N-CNTs(22.85). | ||
The C 1s XPS profile (Fig. 3B) of all supports shows a sharp peak at around 284.6 eV, which belongs to the sp2-hybridized graphitic carbon atoms, as well as a small signal according to C–N species at around 288.5 eV. In the C 1s XPS spectra of N-CNTs, the main peak shifts to a higher binding energy (BE), and becomes wide and asymmetrical. All these changes are due to the disordering of the graphite-like structure after the incorporation of N atoms and the formation of C–N species, which indicates that the incorporation of N presents certain influence on the graphitization and electronic structure of CNTs.
The N 1s XPS profile of N-CNTs in Fig. 3C is broken down into three contributing peaks and analyzed to determine the type of N functionalities present in the samples. The peaks located at 398.7, 399.5, and 400.8 eV are assigned to pyridinic nitrogen (NP), pyrrolic nitrogen (NPYR), and quaternary nitrogen (NQ), respectively. The alteration of the electronic structure in N-dopants strongly depends on bonding configurations. In addition, among these kinds of N atoms, NP is of considerable importance because of the localized electron pair that the atom owned. Primarily, given the larger overlap of the d-orbital of transition metal with the nonbonding state of lone pair electrons, Pt atoms strongly interact with NP at the surface of catalyst. Then, the NP atoms donate electrons to the Pt surface and facilitate the transformation of surface Pt sites into catalytically active sites.34 In addition, the strong interaction of NP and Pt atoms can explain the smaller Pt particles after the incorporation of N.43 Meanwhile, the incorporation of N favors the activation of molecular oxygen by donating an excess electron charge to adsorbed oxygen, generating anionic O2 like superoxo or peroxo oxygen. In addition, NP is bonded heterogeneously to two adjacent carbon atoms with two valence electrons of N in the graphene lattice of CNTs, causing structural deformations and exposing planar edges or defect sites in N-CNTs, which promote the adsorption of glycerol and oxygen. The NP content in the N-CNTs is calculated and presented in Fig. 3C, displaying that the content of NP increases (from 0.22 to 0.39) with the total N content. Thus, we can speculate that the amount of defect sites will increase with N content.
To determine the degree of structural deformations present in N-CNTs, Raman spectroscopy measurement (Fig. 4) is performed. The D band at approximately 1341 cm−1 corresponded to the structural defects on the graphitic plane of CNTs. The G band at approximately 1571 cm−1 is attributed to the E2g vibrational mode present in sp2-bonded graphitic carbons. Furthermore, the intensity ratio of the D and G bands, namely, the ID/IG ratio, indicates the degree of structural defects in the catalyst support materials.45 Higher ratio means more defects on the surface of the carrier. The ID/IG ratio of CNTs-353 is 0.84 and then increases to 1.02 with increasing N content, thus indicating the amount of defect sites increases with N content. This conclusion is in accordance with the results obtained from XPS characterization, and the defect sites avail of the immobilization of metal nanoparticles on the support surface.
 | ||
| Fig. 4 Raman spectra of N-CNTs with different N content. (a) CNTs-353, (b) N-CNTs(3.32), (c) N-CNTs(5.74), and (d) N-CNTs(9.44). | ||
To evaluate the surface acidity/basicity of the supports, TPD-MS (Fig. 5) is conducted. Different surface oxygen-containing groups can be decomposed to CO and/or CO2 at different temperatures. In the literature report,30 CO2 results from the decomposition of carboxylic acids at 150 °C to 450 °C, carboxylic anhydrides at 400 °C to 650 °C, and lactones at 600 °C to 800 °C. CO can originate from the decomposition of carboxylic anhydrides at 400 °C to 650 °C, phenols at 600 °C to 800 °C, and carbonyls/quinones at 750 °C to 1000 °C. From the peaks observed in Fig. 5, the sample oxidized with nitric acid showed the highest amount of surface oxygen from carboxylic acids, which is detrimental for reactant absorption, and then damaged the glycerol oxidation reactions. However, after the incorporation of N, a substantial reduction occurs in carboxylic acid groups. Meanwhile, evident peaks belonging to the degradation of lactone and/or phenol groups are shown on the TPD curves. In the published paper about the oxidation of 5-hydroxymethyl-furfural to 2,5-furandicarboxylic acid in water,46 the authors speculated that the carboxyl groups on CNTs perform a negative function in the catalytic reaction, whereas carbonyl/quinone and/or phenol groups facilitate the conversion of the reactant to intermediate products. Thus, the presence of lactone and phenol groups on the surface of supports after the incorporation of N may perform a positive function in glycerol oxidation. More importantly, the ratio of CO/CO2 can be considered as an indirect measure of surface acidity (low values) or basicity (high values). The ratio of CO/CO2 increases until N content reaches 5.74% and then decreases with further increase in N content, declaring that the highest surface basicity when the N content is 5.74%. The enhanced surface basicity accelerates alkoxide formation by abstracting proton from glycerol in the base-free aqueous solution. Furthermore, lactones and phenol groups on the surface of support materials may promote the absorption of glycerol, as well.
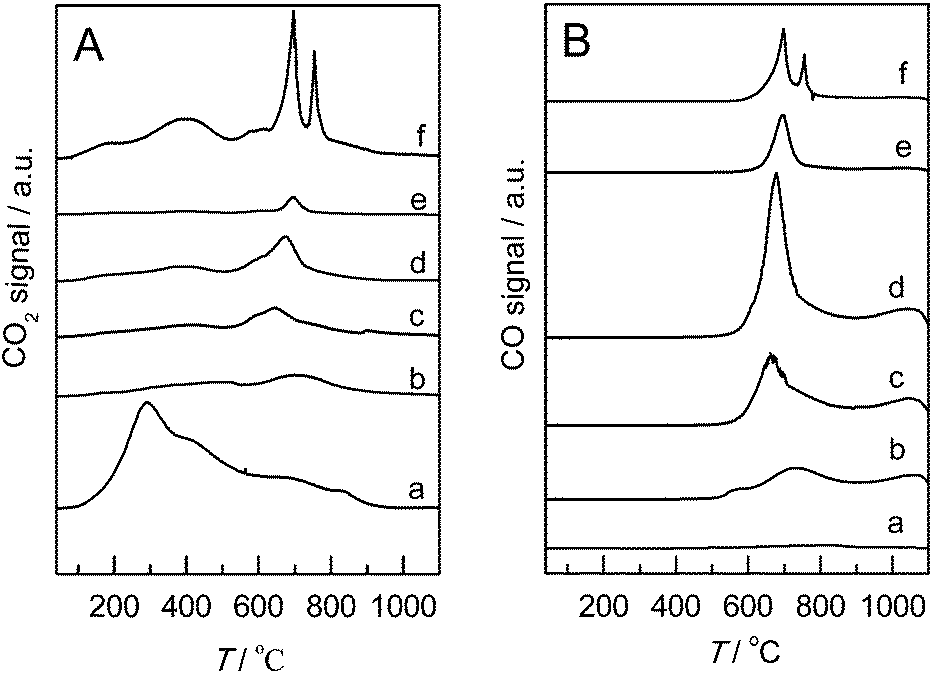 | ||
| Fig. 5 TPD evolution profiles of the different supports (A): CO2, (B): CO, (a) CNTs-353, (b) N-CNTs(1.57), (c) N-CNTs(3.32), (d) N-CNTs(5.74), (e) N-CNTs(9.44), (f) N-CNTs(22.85). | ||
The metal dispersion listed in Table 1 is calculated by TEM characterization and O2–H2 titration results. As seen from the table, the influence of doped N on the particle size and the dispersion of Pt are considerable. D(O2) presents a decreasing trend (from 55.6 to 13.3) along with the increase in N content (from 1.57% to 22.85%). As presented previously, the doping of N atoms favors the absorption of molecular oxygen. Thus, molecular oxygen dissociation is confined, which decreases the chemical adsorption of oxygen atoms. Otherwise, the surface electronegativity of the catalysts after N doping facilitates the chemical adsorption of hydrogen atoms. On the contrary, the chemical adsorption of hydrogen atoms used for reducing the Pt–O species decreases as the chemical adsorption of oxygen atoms is reduced. These two factors make the chemical adsorption of hydrogen atoms initially increase and then decreases with increasing N content. Therefore, D(H2) initially increases and then decreases.
Glycerol oxidation
Catalytic results on catalysts with different supports for aerobic glycerol oxidation under atmospheric pressure and at 60 °C are summarized in Table S2.† Compared with oxide supports, carbonous supports, except C3N4, exhibit relatively good performance, which is probably due to their large specific surface area and excellent electron conductivity. The inferior activity of 1% Pt/C3N4 may be ascribed to smaller specific surface area, which limits the access of reactant to the catalytic active sites. On the other hand, the instability of several oxide supports like TiO2, hydrotalcite, and mesoporous silica in acidic/alkaline conditions restricts their application in glycerol oxidation reactions. As can be seen from the XRD patterns (Fig. S2†) of the catalysts, the Pt particles are loaded on the supports in small sizes and an even dispersion. Thus, we consider that the Pt particle size is not the factor that affects catalytic activity. Instead, the structure and properties of catalysts perform a pivotal function on this property.The catalytic activity of catalysts with different N content in the same reaction condition is presented in Table 2. On 1% Pt/CNTs-353 catalyst, the conversion of glycerol is low (51.5%), and GLYA is the main product (with 43.2% selectivity). The low activity of 1% Pt/CNTs-353 catalyst is due to the large amount of carboxyl groups on the surface of the support after oxidation treatment with nitric acid, which is unfavorable for the adsorption of glycerol on the catalyst surface. After the incorporation of N, the conversion of glycerol is initially enhanced to 76.1%. At the same time, selectivity increases from 43.2% to 55.6% on 1% Pt/N-CNTs(5.74) catalyst. Then, conversion and selectivity are decreased to 2.5% and 20.9%, respectively, with further increase of N content (to 61.67%). The variation trend of TOF agrees with the effect of N content on the glycerol conversion. The TOF of the best performance catalyst (1% Pt/N-CNTs(5.74), 459.2 h−1) is almost twice as high as that of without N doped one (1% Pt/CNTs-353, 248.3 h−1), indicating that the moderate N content has a positive effect on the selective oxidation of glycerol in the base-free aqueous solution. The superior activity after the incorporation of N can be summed up in several aspects: the transfer of electron from NP to Pt facilitates the conversion of surface Pt sites into catalytically active sites. At the same time, the activation of molecular oxygen is accelerated by the donation of an excess electron charge to adsorbed oxygen; the increase in surface defect sites promotes the adsorption of glycerol and oxygen; the enhancement of surface basicity favors the abstraction of proton from glycerol to form the alkoxide. The poor activity of catalysts with higher N content is due to the decrease in specific surface area, which restricts the contact of reactants with the catalytic active sites.
| Catalyst | Conversion/% | Selectivity/% | TOF/h−1 | ||
|---|---|---|---|---|---|
| GLYA | GLYDE | DHA | |||
| a Reaction conditions: 10 mL glycerol aqueous solution (0.1 mol L−1), 0.039 g catalyst, glycerol/Pt = 500 (molar ratio), T = 333 K, P(O2) atmospheric pressure, O2 10 mL min−1, reaction time = 4 h. | |||||
| 1% Pt/CNTs-353 | 51.5 | 43.2 | 26.4 | 23.2 | 248.3 |
| 1% Pt/N-CNTs(1.57) | 49.0 | 48.3 | 19.4 | 25.8 | 249.2 |
| 1% Pt/N-CNTs(3.32) | 58.5 | 47.6 | 19.5 | 24.6 | 312.7 |
| 1% Pt/N-CNTs(5.74) | 76.1 | 55.6 | 19.0 | 20.3 | 459.2 |
| 1% Pt/N-CNTs(9.44) | 14.5 | 40.7 | 29.3 | 20.4 | 152.4 |
| 1% Pt/N-CNTs(22.85) | 7.9 | 32.5 | 33.5 | 20.6 | 74.3 |
| 1% Pt/C3N4(61.67) | 2.5 | 20.9 | 55.1 | 24.0 | — |
The selectivity to GLYA increases with glycerol conversion, whereas the selectivity to DHA remains nearly constant as a function of reaction time. The reason for this finding is that, in base-free conditions, molecules with secondary and primary C–OH groups can be absorbed on Pt atoms on the catalyst surface and then activated. GLYDE and DHA are the two primary products of glycerol oxidation. However, GLYDE is easily oxidized to GLYA, whereas DHA is stable in base-free aqueous solutions.47 Thus, when glycerol conversion increases, selectivity toward GLYDE decreases with the formation of GLYA. In contrast, selectivity toward DHA does not vary significantly.
Catalyst regeneration
To examine the stability of 1% Pt/CNTs-353 and 1% Pt/N-CNTs(5.74) catalysts, recycling experiments are conducted in the same reaction conditions as above (Fig. S3†). After 4 h of reaction, the catalyst is centrifuged, washed with distilled water, and dried at 110 °C before the next round of reaction. After five runs of reaction, glycerol conversion sharply decreases from 76.1% to 29.5% on 1% Pt/N-CNTs(5.74) catalyst. However, on 1% Pt/CNTs-353 catalyst, glycerol conversion during the first four runs exhibits a slight variation. The morphology of the recycled catalysts is investigated by XRD (Fig. S4†) and TEM (Fig. S5†). Results declare that, after reactions, Pt particles slightly increase from 2.7 to 3.3 nm and from 2.3 to 3.2 nm for 1% Pt/CNTs-353 and 1% Pt/N-CNTs(5.74) catalysts, respectively. Nevertheless, with prolonged reaction time to 12 h on 1% Pt/CNTs-353 catalyst, glycerol conversion decreases sharply from 93.4% (cycle 1) to 66.6% (cycle 4). This decline may be due to the main disadvantage of catalysts based on Pt-groups, namely, their deactivation at increasing reaction time because of oxygen poisoning. According to the literature,48 the lifetime and activity of Pt-based catalysts are limited by, at least, the following three processes: irreversible oxidation of metallic Pt to PtOx by dioxygen; increase in Pt particle size during reaction, and; irreversible adsorption of acid products on the surface of catalysts. To determine which factor presents the vital influence on catalyst deactivation, another 1% Pt/CNTs-353 catalyst with a larger particle size (5.3 nm from XRD) is produced and compared with the previous catalyst (2.7 nm from TEM). As a result, Pt particles with 5.3 nm diameter exhibits similar behavior (with a 49.1% conversion), which agrees with the results of a published paper stating that the conversion of glycerol is similar when the particle size of Pt is smaller than 6.0 nm.10 Therefore, we can conclude that the impact of particle size is negligible on the deactivation of Pt-based catalysts tested in this work.Next, TGA is performed to verify the irreversible adsorption of acidic products on the catalyst surface, which contribute to catalyst deactivation. TGA results of fresh and recycled catalysts are plotted in Fig. S6.† Below 150 °C, the slight weight loss observed for all samples corresponds to the evaporation of physically adsorbed water. Then, the minor weight loss below 500 °C is attributed to the decarboxylation and elimination of hydroxyl functionalities. Subsequently, the drastic weight loss occurring over 500 °C is caused by the degradation of disordered carbon. Comparing fresh catalysts with catalysts obtained after five reaction runs, we find that the absorption of acid products during reaction is not significant, thereby exhibiting that the absorption of acid products is not the key factor to catalyst deactivation. Therefore, we assert that oxygen poisoning of Pt-based catalysts is the fatal factor for its deactivation. Combining the results of XPS and Raman analysis, in which more structure defects are formed because of N incorporation, we predict that the deactivation process will be accelerated on 1% Pt/N-CNTs(5.74) catalyst as the absorption of oxygen is promoted.
Given that the deactivation essence of Pt-based catalysts is the irreversible oxidation of active Pt nanoparticles to inert PtOx by dioxygen, PtOx must be reduced to metallic Pt nanoparticles to regenerate the catalyst. Then, after reaction, the catalyst is filtered, and washed with distilled water to remove the absorbed acid products and other impurities. After drying in an oven at 110 °C, catalytic power is reduced in H2 following 230 °C for 3 h. Finally, the powder is applied in the next reaction after cooling to room temperature.
The activity of regenerated 1% Pt/N-CNTs(3.32) catalyst is displayed in Fig. 6. After reaction, when the catalyst is treated with hydrogen, both glycerol conversion and GLYA selectivity can remain nearly constant in the recycling reaction, indicating the complete regeneration of the catalyst after oxygen poisoning. This finding is in accordance with the previous conclusion that oxygen poisoning of Pt-based catalysts is the fatal factor for deactivation, and that surface active Pt nanoparticles is oxidized to the inert PtOx after oxygen poisoning.
 | ||
| Fig. 6 Activity of the 1% Pt/N-CNTs(3.32) catalyst after regeneration. | ||
Catalytic mechanism
Based on the above results, N-CNTs is a preferable support material for the aerobic oxidation of glycerol. It has been reported that the anionic metal plays an important role in adsorbing O2 on the metal surface and the activation of molecular oxygen by donating an excess electron charge to the adsorbed oxygen generates the anionic O2 like superoxo or peroxo oxygen, thereby the catalytic activity for the oxidation of polyols is enhanced.49,50 Taking the reports in literature and the experiments at present into account, we propose a plausible reaction mechanism of glycerol oxidation in the base-free aqueous solution over 1% Pt/N-CNTs(x) catalyst as shown in Scheme 1. | ||
| Scheme 1 Proposed reaction pathway for glycerol oxidation catalyzed by Pt/N-CNTs. | ||
Initially, O2 is adsorbed onto the surface sites of Pt atoms. Electron transfer from NP to Pt favors the activation of molecular oxygen by donating an excess electron charge to the adsorbed oxygen.51–54 After that, the hydroxy proton is abstracted to form the alkoxide, enhancing the binding of glycerol to metal atoms. Then, the proton on α-CH2 is transferred from the adsorbed alkoxide carbon to the absorbed oxygen, releasing GLYDE and H2O to generate back anionic O2− species. Next, GLYDE is attracted by the adjacent Pt atom with adsorbed oxygen, creating a partial positive charge on the carbonyl carbon. Thus, the nucleophilic oxygen atom of water attacks the electron-deficient carbonyl carbon. Finally, the adsorbed oxygen abstracts the proton from H2O, yielding GLYA, and is then changed back to peroxo form to complete the catalytic cycle.
4. Conclusions
N-CNTs with different N-content are synthesized by annealing the mixture of CNTs and melamine with different mass ratios. Pt/N-CNTs with high dispersion of Pt nanoparticles are prepared by wet impregnation method, followed by liquid-phase and gas reductions. Higher catalytic performance for glycerol oxidation in terms of conversion and GLYA selectivity is obtained over Pt/N-CNTs than over Pt/CNTs with molecular oxygen in base-free aqueous solutions. Glycerol conversion is enhanced from 51.5% on 1% Pt/CNTs to 76.1% on 1% Pt/N-CNTs, with a corresponding increase in the selectivity to GLYA from 44.1% to 55.6% at 60 °C for 4 h. This enhancement is ascribed to the increased surface basicity that originates from the incorporated N-atoms in Pt/N-CNTs, which accelerates the activation of –OH groups in glycerol to alkoxide. The 1% Pt/N-CNTs can be reused for several times with negligible changes in structure and performance when the used catalyst is reduced in H2 atmosphere at 230 °C for 3 h.Acknowledgements
We acknowledge the financial support from the National Basic Research Program of China (no. 2011CBA00508), the Natural Science Foundation of China (nos 21303141, 21403178 and 21473145), the Research Fund for the Doctoral Program of Higher Education (no. 20110121130002), and the Program for Innovative Research Team in Chinese Universities (no. IRT_14R31).Notes and references
- M. Höök and X. Tang, Energy Policy, 2013, 52, 797–809 CrossRef PubMed.
- A. Demirbas, Energy Convers. Manage., 2009, 50, 14–34 CrossRef CAS PubMed.
- I. Sobczak, K. Jagodzinska and M. Ziolek, Catal. Today, 2010, 158, 121–129 CrossRef CAS PubMed.
- S. Gil, L. Muñoz, L. Sánchez-Silva, A. Romero and J. L. Valverde, Chem. Eng. J., 2011, 172, 418–429 CrossRef CAS PubMed.
- B. Katryniok, H. Kimura, E. Skrzynska, J.-S. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Green Chem., 2011, 13, 1960–1979 RSC.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- C. L. Bianchi, P. Canton, N. Dimitratos, F. Porta and L. Prati, Catal. Today, 2005, 102–103, 203–212 CrossRef CAS PubMed.
- N. Dimitratos, F. Porta and L. Prati, Appl. Catal., A, 2005, 291, 210–214 CrossRef CAS PubMed.
- N. Dimitratos, C. Messi, F. Porta, L. Prati and A. Villa, J. Mol. Catal. A: Chem., 2006, 256, 21–28 CrossRef CAS PubMed.
- D. Liang, J. Gao, J. Wang, P. Chen, Z. Hou and X. Zheng, Catal. Commun., 2009, 10, 1586–1590 CrossRef CAS PubMed.
- N. Dimitratos, A. Villa and L. Prati, Catal. Lett., 2009, 133, 334–340 CrossRef CAS PubMed.
- R. Garcia, M. Besson and P. Gallezot, Appl. Catal., A, 1995, 127, 165–176 CrossRef CAS.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin and G. J. Hutchings, Chem. Commun., 2002, 696–697 RSC.
- S. Gil, M. Marchena, L. Sánchez-Silva, A. Romero, P. Sánchez and J. L. Valverde, Chem. Eng. J., 2011, 178, 423–435 CrossRef CAS PubMed.
- E. G. Rodrigues, S. A. C. Carabineiro, J. J. Delgado, X. Chen, M. F. R. Pereira and J. J. M. Órfão, J. Catal., 2012, 285, 83–91 CrossRef CAS PubMed.
- S. Gil, P. J. Lucas, A. Nieto-Márquez, L. Sánchez-Silva, A. Giroir-Fendler, A. Romero and J. L. Valverde, Ind. Eng. Chem. Res., 2014, 53, 16696–16706 CrossRef CAS.
- F. Porta and L. Prati, J. Catal., 2004, 224, 397–403 CrossRef CAS PubMed.
- N. Dimitratos, J. Lopez-Sanchez, D. Lennon, F. Porta, L. Prati and A. Villa, Catal. Lett., 2006, 108, 147–153 CrossRef CAS PubMed.
- J. Gao, D. Liang, P. Chen, Z. Hou and X. Zheng, Catal. Lett., 2009, 130, 185–191 CrossRef CAS.
- D. Liang, J. Gao, H. Sun, P. Chen, Z. Hou and X. Zheng, Appl. Catal., B, 2011, 106, 423–432 CrossRef CAS PubMed.
- N. Mimura, N. Hiyoshi, T. Fujitani and F. Dumeignil, RSC Adv., 2014, 4, 33416–33423 RSC.
- L. Prati, A. Villa, C. E. Chan-Thaw, R. Arrigo, D. Wang and D. S. Su, Faraday Discuss., 2011, 152, 353–365 RSC.
- N. Dimitratos, A. Villa, D. Wang, F. Porta, D. Su and L. Prati, J. Catal., 2006, 244, 113–121 CrossRef CAS PubMed.
- A. Tsuji, K. T. V. Rao, S. Nishimura, A. Takagaki and K. Ebitani, ChemSusChem, 2011, 4, 542–548 CrossRef CAS PubMed.
- D. Liang, J. Gao, J. Wang, P. Chen, Y. Wei and Z. Hou, Catal. Commun., 2011, 12, 1059–1062 CrossRef CAS PubMed.
- D. Tongsakul, S. Nishimura, C. Thammacharoen, S. Ekgasit and K. Ebitani, Ind. Eng. Chem. Res., 2012, 51, 16182–16187 CrossRef CAS.
- X. Wang, G. Wu, F. Wang, K. Ding, F. Zhang, X. Liu and Y. Xue, Catal. Commun., 2012, 28, 73–76 CrossRef CAS PubMed.
- A. Iriondo, J. F. Cambra, V. L. Barrio, M. B. Guemez, P. L. Arias, M. C. Sanchez-Sanchez, R. M. Navarro and J. L. G. Fierro, Appl. Catal., B, 2011, 106, 83–93 CAS.
- G. L. Brett, Q. He, C. Hammond, P. J. Miedziak, N. Dimitratos, M. Sankar, A. A. Herzing, M. Conte, J. A. Lopez-Sanchez, C. J. Kiely, D. W. Knight, S. H. Taylor and G. J. Hutchings, Angew. Chem., Int. Ed., 2011, 50, 10136–10139 CrossRef CAS PubMed.
- E. G. Rodrigues, J. J. Delgado, X. Chen, M. F. R. Pereira and J. J. M. Órfão, Ind. Eng. Chem. Res., 2012, 51, 15884–15894 CrossRef CAS.
- R. Lv, T. Cui, M.-S. Jun, Q. Zhang, A. Cao, D. S. Su, Z. Zhang, S.-H. Yoon, J. Miyawaki, I. Mochida and F. Kang, Adv. Funct. Mater., 2011, 21, 999–1006 CrossRef CAS PubMed.
- R. Arrigo, M. Hävecker, S. Wrabetz, R. Blume, M. Lerch, J. McGregor, E. P. J. Parrott, J. A. Zeitler, L. F. Gladden, A. Knop-Gericke, R. Schlögl and D. S. Su, J. Am. Chem. Soc., 2010, 132, 9616–9630 CrossRef CAS PubMed.
- D. C. Higgins, D. Meza and Z. Chen, J. Phys. Chem. C, 2010, 114, 21982–21988 CAS.
- I. Florea, O. Ersen, R. Arenal, D. Ihiawakrim, C. Messaoudi, K. Chizari, I. Janowska and C. Pham-Huu, J. Am. Chem. Soc., 2012, 134, 9672–9680 CrossRef CAS PubMed.
- R. Czerw, M. Terrones, J. C. Charlier, X. Blase, B. Foley, R. Kamalakaran, N. Grobert, H. Terrones, D. Tekleab, P. M. Ajayan, W. Blau, M. Rühle and D. L. Carroll, Nano Lett., 2001, 1, 457–460 CrossRef CAS.
- T. Zhou, H. Wang, J. Key, S. Ji, V. Linkov and R. Wang, RSC Adv., 2013, 3, 16949–16953 RSC.
- Y. Wang, X. Wang, M. Antonietti and Y. Zhang, ChemSusChem, 2010, 3, 435–439 CrossRef CAS PubMed.
- Y. Cao, H. Yu, F. Peng and H. Wang, ACS Catal., 2014, 4, 1617–1625 CrossRef CAS.
- Y. T. Lee, N. S. Kim, S. Y. Bae, J. Park, S.-C. Yu, H. Ryu and H. J. Lee, J. Phys. Chem. B, 2003, 107, 12958–12963 CrossRef CAS.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- C. Wang, G. Yi, H. Lin and Y. Yuan, Int. J. Hydrogen Energy, 2012, 37, 14124–14132 CrossRef CAS PubMed.
- Y. Shao, J. Sui, G. Yin and Y. Gao, Appl. Catal., B, 2008, 79, 89–99 CrossRef CAS PubMed.
- C. E. Chan-Thaw, A. Villa, L. Prati and A. Thomas, Chem.–Eur. J., 2011, 17, 1052–1057 CrossRef CAS PubMed.
- C.-L. Sun, C.-W. Pao, H.-M. Tsai, J.-W. Chiou, S. C. Ray, H.-W. Wang, M. Hayashi, L.-C. Chen, H.-J. Lin, J.-F. Lee, L. Chang, M.-H. Tsai, K.-H. Chen and W.-F. Pong, Nanoscale, 2013, 5, 6812–6818 RSC.
- X. Wan, C. Zhou, J. Chen, W. Deng, Q. Zhang, Y. Yang and Y. Wang, ACS Catal., 2014, 4, 2175–2185 CAS.
- E. G. Rodrigues, M. F. R. Pereira, J. J. Delgado, X. Chen and J. J. M. Órfão, Catal. Commun., 2011, 16, 64–69 CrossRef CAS PubMed.
- J. W. Nicoletti and G. M. Whitesides, J. Phys. Chem., 1989, 93, 759–767 CrossRef CAS.
- D. Tongsakul, S. Nishimura and K. Ebitani, ACS Catal., 2013, 3, 2199–2207 CAS.
- D. Tongsakul, S. Nishimura and K. Ebitani, J. Phys. Chem. C, 2014, 118, 11723–11730 CAS.
- M. Okumura, Y. Kitagawa, T. Kawakami and M. Haruta, Chem. Phys. Lett., 2008, 459, 133–136 CrossRef CAS PubMed.
- M. Okumura, Y. Kitagawa, M. Haruta and K. Yamaguchi, Appl. Catal., A, 2005, 291, 37–44 CrossRef CAS PubMed.
- N. K. Chaki, H. Tsunoyama, Y. Negishi, H. Sakurai and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 4885–4888 CAS.
- Y. S. Kim, A. Bostwick, E. Rotenberg, P. N. Ross, S. C. Hong and B. S. Mun, J. Chem. Phys., 2010, 133, 034501 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Physical properties of the catalyst supports. TEM images of catalysts 1% Pt/CNTs-353 and 1% Pt/N-CNTs(x). XRD patterns of the catalysts with different support materials. TEM images of catalysts after 5 runs of reaction and XRD patterns of these catalysts before and after reaction. TGA curves of catalysts before and after 5 runs reactions. The activity of 1% Pt/CNTs-353 and 1% Pt/N-CNTs(5.74) catalysts in recycling experiments. See DOI: 10.1039/c5ra02112j |
| This journal is © The Royal Society of Chemistry 2015 |