
Assembly of anisotropic one dimensional Ag nanostructures through orientated attachment: on-axis or off-axis growth?†
Weiqiang Lva,
Yaxing Zhua,
Yinghua Niua,
Weirong Huoa,
Kang Lia,
Gaolong Zhua,
Yachun Lianga,
Wenzhan Wub and
Weidong He*acd
aSchool of Energy Science and Engineering, University of Electronic Science and Technology of China (UESTC), Chengdu 611731, PR China. E-mail: weidong.he@uestc.edu.cn; weidong.he@vanderbilt.edu
bDepartment of Chemistry, The Hong Kong University of Science and Technology, Hong Kong SAR, PR China
cInterdisciplinary Program in Materials Science, Vanderbilt University, Nashville, Tennessee 37234-0106, USA
dVanderbilt Institute of Nanoscale Science and Engineering, Vanderbilt University, Nashville, Tennessee 37234-0106, USA
First published on 17th February 2015
Abstract
Recently it has been found that one dimensional (1D) Ag nano-structures can be synthesized through oriented attachment (OA) growth. In OA growth of 1D structures, nanoparticles (NPs) attach to the growing nanorods (NRs) via either on-axis or off-axis attachment. However, the thermodynamic basis for understanding the preference of each growth mode has remained unexplored until now. In this paper, molecular static calculations are performed to investigate the van der Waals interactions (vdW) in both on-axis and off-axis attachments of 1D Ag nano-structures. The correlation of parameters including the size, aspect ratio (AR), crystalline orientation of NR, the inter-particle separation and the off-axis approaching angle, with both OA attachments is investigated in detail. The results show that off-axis attachment is thermodynamically favorable compared to on-axis attachment in a typical OA growth, and straight on-axis OA growth are typically realized by tuning the other aspects of an OA growth system. Interestingly, it is found that the off-axis growth is both precursor-size dependent and crystalline-orientation dependent.
1. Introduction
One dimensional (1D) anisotropic Ag nanostructures exhibit excellent electric, optical and catalytic properties and can be applied in many fields such as transistors, optoelectronics, surface-enhanced Raman spectroscopy (SERS), and catalysis.1–4 Synthesis of structurally-well defined 1D Ag nanostructures with a homogenous size distribution largely facilitates their applications in these areas. Much effort has been focused on synthesizing Ag nanorods (NRs)/nanowires (NWs) with various sizes and aspect ratios (ARs).5–8 In particular, high-quality 1D Ag nano-structures have been synthesized through oriented attachment (OA) growth.9–12 In the OA growth, different from the traditional Ostwald ripening (OR) growth, larger nanocrystals grow through the attachment of two primary nanoparticles directly along specific crystalline orientations.13–15 OA has shown great advantages in synthesizing nanostructures of various materials.16–19 However, realizing nanostructures with desired morphology and size control remains challenging in the general OA field. Towards this direction, an insightful understanding of the nanocrystal growth mechanism facilitates the efforts for the rational design of an efficient OA growth system.At present, there are two research trajectories of investigating OA growth mechanism. One trajectory is to monitor nanoparticle (NP) motions by in situ HR-TEM techniques and to extract nanoscale forces behind these motions.20–22 The other research trajectory is to carry out computer simulations by building various growth models.23–25 For instance, Sathiyanarayanan et al. found that solvent plays a dominant role in the anisotropic growth of colloidal Ag nano-structures.26 The authors studied vdW and Coulombic interaction (CI) involved in OA growth of NRs by constructing mathematical models. The correlation of parameters including the size and aspect ratio of NRs, with the OA growth has been investigated from an energy point of view.27–31 Although these analytic results facilitate the understanding of OA thermodynamics and kinetics, much regarding the OA growth thermodynamics still remains elusive. For instance, it has been observed experimentally that NPs can attach NRs either through on-axis attachment to form a straight NRs or through off-axis growth to result in zigzag or curly 1D structures and 3D fractal structures.9–12,32,33 The different Ag nano-structures are known to exhibit dramatically-different physical properties, and thus the fundamental understanding and rational design over such nano-structures are of particular significance to the OA field. Unfortunately, the thermodynamic origins of both on-axis and off-axis growth mechanisms have not been well understood till this point. In this paper, molecular static calculations are employed to investigate the van der Waals interactions (vdW) in the OA assembly of both on axis and off-axis attachments since vdW is a fundamental attractive driving force and in some cases even dominates the OA growth of a variety of nanostructures.34–36 The important parameters associated with OA growth, such as NR diameter, aspect ratio and crystalline orientation, are incorporated in our calculations. The theoretical calculation and analysis facilitate the understanding of OA assembly mechanism of anisotropic 1D materials and the rational pre-experiment design of synthetic parameters.
2. Models and methods
Fig. 1 shows NR–NP assembly models employed in the molecule static calculation. An NP with a size of a × a × a approaches the growing NR with a length of l, a cross section of a × a and a crystalline orientation of [hkl] along the NR axis direction. The aspect ratio (AR) of the NR can then be defined as l/a and d is the surface-to-surface inter-particle separation between the NR and the NP. Fig. 1a shows that the NP approaches the NR through on-axis assembly, while in Fig. 1b the NP approaches the NR through off-axis assembly with an approaching angle of θ.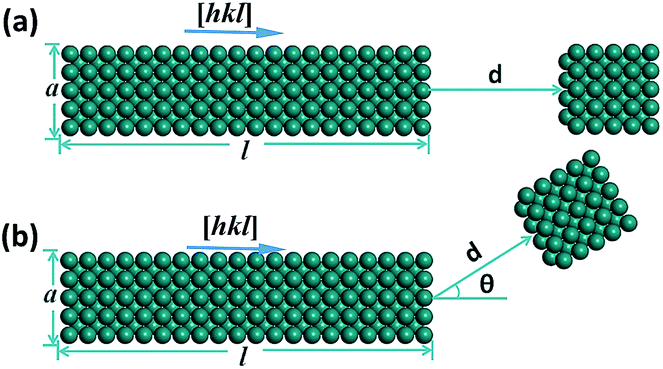 | ||
| Fig. 1 NR–NP attachment modes of (a) on-axis growth, and (b) off-axis growth. A nano-cube with a size of a × a × a stands for an NP. A nano-rectangular with a length of l and a cross-section of a × a stands for an NR. AR = l/a and [hkl] is the crystalline direction. d is the surface-to-surface inter-particle separation between the NR and the NP, and θ is the approaching angle of an NP towards an NR. | ||
vdW is then calculated by molecular static simulation. The universal force field (UFF) is employed,37 in which the vdW are described by the Lennard-Jones potential given in eqn (1),38
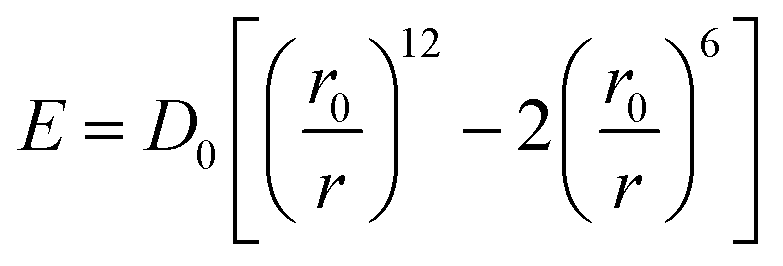 | (1) |
| ΔEvdw(d) = Evdw(d) − Evdw(∞) | (2) |
3. Results and discussions
It is well known that varying the diameters of Ag NRs can tune their optical properties; for instance, the surface plasmon resonance peak can be shifted from visible-light ranges to near infrared regime.39 Growth of NRs with different diameters is of great significance and the growth mechanism deserves fundamental investigation. A small diameter range from 0.4 nm to 4 nm for Ag NP precursors is selected for the simulation according to the typical experimental values.10,35 No larger NP diameter is chosen since the rate constant of OA growth decreases exponentially as NP size increases.40 As shown in Fig. 1, vdW associated with the on-axis OA growth of NRs with different diameters and along three different crystalline orientations, is studied. As the NR–NP separation increases, vdW drops rapidly for NRs and NPs with all four different diameters varying from 0.4 nm to 4 nm. There exists a critical separation, larger than which the decrease of vdW becomes rather gradual. This critical separation for NRs and NPs in the diameter range of 0.4–4 nm is approximately 0.5 nm. In the NR diameter range investigated here, OA assembly along [111] crystalline orientation experiences smaller vdW compared with [100] and [110] directions. Such a difference is more obvious for OA growth of NRs with smaller diameters. For instance, at zero separation, the difference of vdW along [110] and [100] is nearly twice the vdW along [111] direction for NRs of 0.4 nm in diameter, as shown in Fig. 2a. While such a difference is approximately 1.3, 1.2 and 1.1 times for NRs of 1.2, 2.5 and 4 nm in diameter, respectively, as shown in Fig. 2b–d. The vdW of OA assembly along [100] and [110] shows a rather small difference as evidenced by the nearly overlapped red curve and black curve. Therefore, Ag NRs growth along [111] crystalline direction experiences smaller attractive vdW and thus is thermodynamically unfavorable, especially for OA growth of NRs with small diameters. The result of crystalline-oriented OA assembly of Ag is consistent with the work of Zhang et al., in which they also show OA growth of Ag along [111] is favored compared to other crystalline directions.23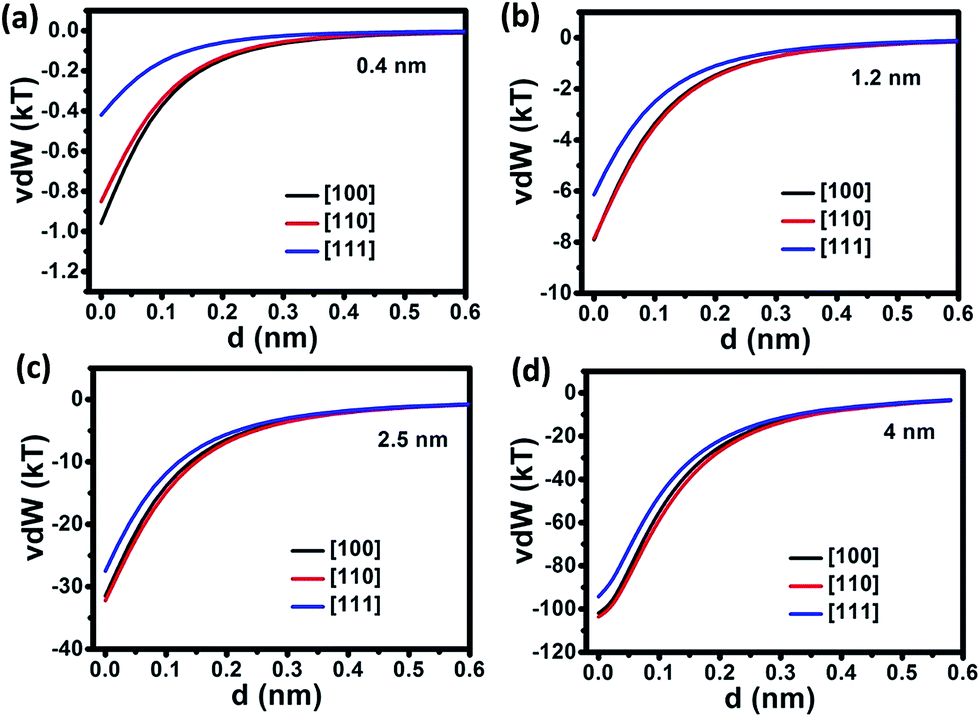 | ||
| Fig. 2 Calculated vdW interaction versus separation between NR and NP of different crystal faces along the axes of NRs with the diameters of (a) 0.4 nm, (b) 1.2 nm, (c) 2.5 nm and (d) 4 nm. The AR of Ag NRs is fixed at 10 and the temperature (T) is 293 K. | ||
In the OA growth of NRs, despite the on-axis assembly of NPs, off-axis growth is also observed in experiments.35,36,41 The vdW in the off-axis OA growth is then investigated, as shown in Fig. 3. With a fixed NR–NP separation, vdW increases as the approaching angle increases in off-axis growth of NRs along all the three different crystalline orientations. Such an increase become more rapid as the approaching angle is over 20°. The result indicates that off-axis OA assembly is favored thermodynamically in a vdW dominated system such as bared or neutral ligand-capped metal and other non-polarized nanoparticle precursors.35,36,41 As the approaching angle in off-axis growth increases, the difference of vdW for different crystalline orientations increases. Such a difference becomes larger as the approaching angle exceeds 30°. The vdW in OA assembly of NRs along different crystalline orientations follows the order of [100] > [110] > [111]. Therefore, the off-axis OA growth is crystalline-orientation dependent. The difference of vdW between off-axis growth and on-axis growth along [100] is the largest among the three crystalline orientations, and off-axis growth along [100] is more preferable. To synthesize [100]-oriented Ag nanorods and nanowires, surface ligands which are strongly attractive to the side-surfaces other than (100) facets should be employed, or specific solvent should be used.10,12,26 Fig. 3b–d shows the plots of vdW versus NR–NP separation in the off-axis growth of OA assembly along three different crystalline orientations, respectively. In all the three considered NR crystalline orientations, the vdW in the off-axis growth decreases significantly as the separation increases. With a fixed separation in the range of 0.8 nm to 1.4 nm, vdW increases as the approaching angle increases. The results indicate that OA growth of un-capped NRs experiences larger thermodynamic driving forces in the off-axis growth, especially in a precursor media with a higher-concentration.
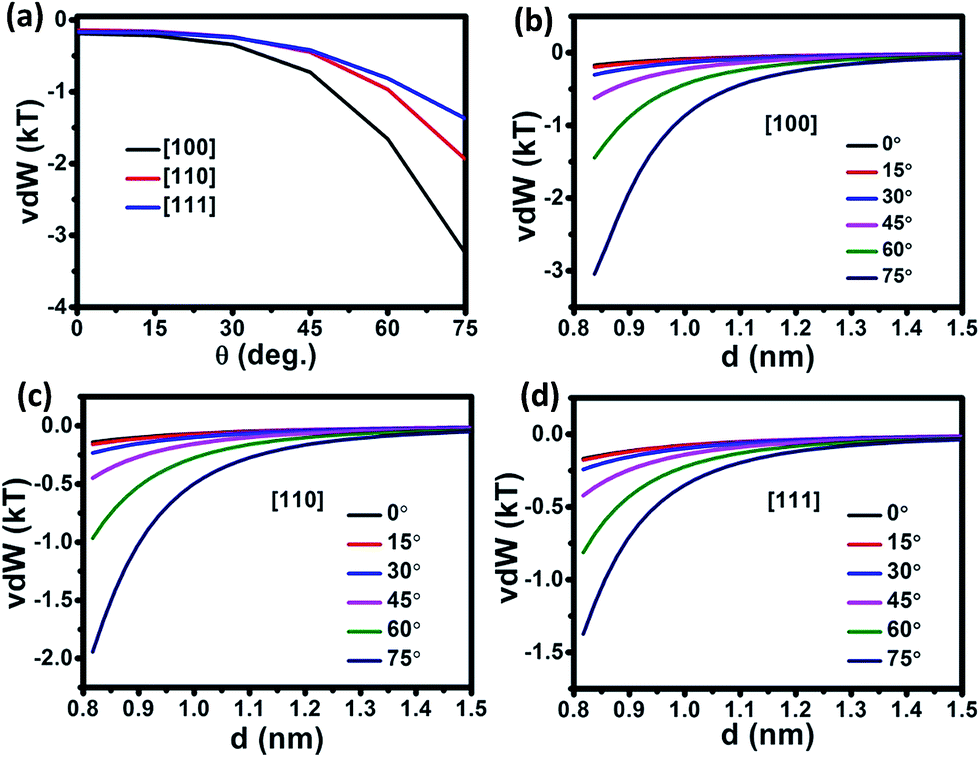 | ||
| Fig. 3 (a) Calculated vdW interaction versus approaching angle of off-axis growth and vdW versus the separation between NR and NP of different angles in the (b) [100], (c) [110] and (d) [111] crystalline growth orientations. The diameter of Ag NR and NP is fixed at 1.2 nm, the AR of NR is 10 and the temperature is 293 K. | ||
vdW involving in the off-axis growth through OA assembly between NRs and NPs with different diameters and ARs is further investigated while fixing OA assembly along the [100] crystalline orientation. As shown in Fig. 4a, vdW increases as NP approaching angle increases, and this increase of vdW becomes much larger as the diameter of NRs increases from 0.4 nm to 4.0 nm. For instance, as shown in the inset of Fig. 4a, the increase of vdW with respect to the approaching angle is rather small for NRs with a diameter smaller than 1.2 nm. The vdW increases approximately 4 and 24 times for 2.5 nm and 4 nm NRs respectively, as the approaching angle increases from 0 to 75°. This result indicates that NRs and NPs with larger diameters are more likely to experience off-axis growth through OA assembly if vdW dominates the OA growth. This conclusion is supported by OA assembly of Cu nanoseeds carried out by Shen et al., since Cu has similar face-centered cubic structure with Ag.42 They demonstrated that 2–3 nm Cu nanoseeds assembled into nanorods or nanodiscs, favor on-axis OA growth, whereas 5 nm Cu nanoseeds assembled into 3D irregular particles, favor off-axis growth. They ascribed the reason to larger inter-particle vdW interaction with increased precursor size as well as the synergistic effect of surface ligands. It should be noted that organic ligands, solvents as well as other synthetic parameters impact largely the OA growth and can be adjusted to tune the ratio of on-axis/off-axis assemblies in the anisotropic growth of various materials including Ag.26,27,36,42,43 To understand the assembly thermodynamics of NRs with different lengths, vdW in the OA growth of NRs with different lengths is evaluated. As shown in Fig. 4b, the five plots of vdW versus θ for NRs with different ARs overlap completely. This indicates that vdW varies little as AR increases, meaning that increasing NR length of NRs has little effect on the thermodynamics of the off-axis OA growth of NRs.
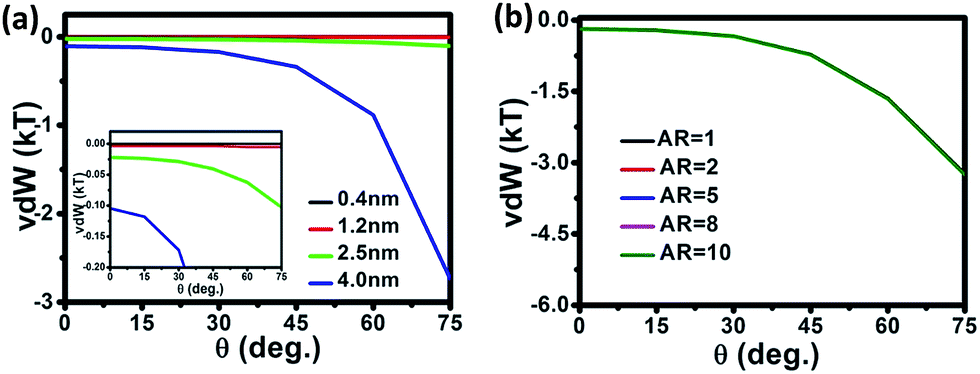 | ||
| Fig. 4 Calculated vdW interaction versus angle between NR and NP of (a) different diameters with the separation fixed at 2.5 nm and AR at 10, and (b) different ARs with the diameter fixed at 1.2 nm and separation at 0.8 nm. The crystalline orientation is [100] and the temperature is 293 K. | ||
In this work, Ag is selected as a materials platform in this study, but the conclusions in this report are applicable to many other materials systems in which vdW dominates the OA growth, such as the frequently reported metal nanoparticles like Au, Cu, Pt, etc. Similar simulations are carried out on these materials and the data are shown in the ESI.† The result indicates that off-axis assemblies of these materials experience larger van der Waals interaction compared with the on-axis assemblies, which is consistent with our conclusion on Ag. In addition, at fixed approaching angle, vdW varies for different materials and follows the order: Au > Pt > Ag > Cu, indicating that the off-axis OA growth is material-dependent. This result may arise from the differences of atomic/crystalline parameters and the Hamaker constants of these metals. Assemblies of nanoparticles with strong polarization, dipoles and surface charges, such as metal oxides and sulfides, are dominated possibly by electrostatic forces rather than vdW and may be thus inconsistent with our result. For instance, the vdW data for the on-axis and off-axis growth of TiO2, ZnO and SnO2 are also shown in the ESI,† illustrating that the off-axis assemblies of these materials experience larger vdW compared with the on-axis assemblies. However, these materials are reported frequently to follow 1D on-axis OA growth.44–46 The calculation of inter-particle electrostatic forces relies on quantitative evaluation of the surface charge distribution of NPs, which requires a combination of first-principle method and molecular dynamics method. Such work will be the future subject of study. Nevertheless, our calculation on vdW facilitates the understanding of the size-dependent on-axis and off-axis OA growth of a variety of materials, including Ag, Au, Cu, Pt, Pd, etc.
4. Conclusions
In summary, the van der Waals interaction associated with on-axis and off-axis orientated attachment growth of 1D anisotropic Ag nanostructure is investigated by molecular static calculations. The results show that the off-axis growth experiences larger vdW and is thermodynamically favorable compared to the on-axis OA growth. Specific ligands, solvents or other synthetic parameters should be adjusted to achieve the anisotropic on-axis OA growth. It is found that the off-axis OA growth is precursor-size dependent. Ag NP precursors of a large size are more likely to grow in an off-axis fashion compared with Ag NPs of a small size. In addition, the off-axis OA growth is crystalline-orientation dependent. Ag NRs growing along the [100] orientation favor more an off-axis growth compared with the growth along the [110] and [111] directions. Our conclusions are applicable to all growth systems in which the OA assembly is mainly governed by vdW interaction, such as neutrally-capped metal nanoparticles. Our work improves our understanding of the OA assembly mechanism of 1D nanocrystals from an energy point of view.Acknowledgements
The work is supported by the UESTC new faculty startup fund and the National Natural Science Foundation of China (grant no. 21403031).References
- Q. Zhou, Y. He, J. Abell, Z. Zhang and Y. Zhao, J. Phys. Chem. C, 2011, 115(29), 14131–14140 CAS.
- T. Y. Kim, W. J. Kim, S. H. Hong, J. E. Kim and K. S. Suh, Angew. Chem., Int. Ed., 2009, 48, 3806–3809 CrossRef CAS PubMed.
- H. Ditlbacher, A. Hohenau, D. Wagner, U. Kreibig, M. Rogers, F. Hofer, F. R. Aussenegg and J. R. Krenn, Phys. Rev. Lett., 2005, 95, 257403 CrossRef.
- Q. Lu, Z. Lu, Y. Lu, L. Lv, Y. Ning, H. Yu, Y. Hou and Y. Yin, Nano Lett., 2013, 13(11), 5698–5702 CrossRef CAS PubMed.
- A. Graff, D. Wagner, H. Ditlbacher and U. Kreibig, Eur. Phys. J. D, 2005, 34, 263–269 CrossRef CAS.
- Y. G. Sun and Y. N. Xia, Adv. Mater., 2002, 14(11), 833–837 CrossRef CAS.
- C. J. Murphy and N. R. Jana, Adv. Mater., 2002, 14(1), 80–82 CrossRef CAS.
- P. Xu, S. H. Jeon, H. T. Chen, H. M. Luo, G. F. Zou, Q. X. Jia, M. Anghel, C. Teuscher, D. J. Williams, B. Zhang, X. J. Han and H. L. Wang, J. Phys. Chem. C, 2010, 114, 22147–22154 CAS.
- H. Liang, D. Rossouw, H. Zhao, S. K. Cushing, H. Shi, A. Korinek, H. Xu, F. Rosei, W. Wang, N. Wu, G. A. Botton and D. Ma, J. Am. Chem. Soc., 2013, 135(26), 9616–9619 CrossRef CAS PubMed.
- M. Giersig, I. Pastoriza-Santos and L. M. Liz-Marzán, J. Mater. Chem., 2004, 14(4), 607–610 RSC.
- H. Liang, H. Zhao, D. Rossouw, W. Wang, H. Xu, G. A. Botton and D. Ma, Chem. Mater., 2012, 24(12), 2339–2346 CrossRef CAS.
- P. Peng, L. Liu, A. P. Gerlich, A. Hu and Y. N. Zhou, Part. Part. Syst. Charact., 2013, 30(5), 420–426 CrossRef CAS.
- R. L. Penn and J. F. Banfield, Science, 1998, 281, 969–971 CrossRef CAS.
- J. Zhang, F. Huang and Z. Lin, Nanoscale, 2010, 2, 2873 RSC.
- Q. Zhang, S. J. Liu and S. H. Yu, J. Mater. Chem., 2009, 19, 191–207 RSC.
- A. Halder and N. Ravishankar, Adv. Mater., 2007, 19(14), 1854–1858 CrossRef CAS.
- H. Y. Liang, H. G. Zhao, D. Rossouw, W. Z. Wang, H. X. Xu, G. A. Botton and D. L. Ma, Chem. Mater., 2012, 24, 2339–2346 CrossRef CAS.
- H. Song, K. H. Lee, H. Jeong, S. H. Um, G. S. Han, H. S. Jung and G. Y. Jung, Nanoscale, 2013, 5, 1188–1194 RSC.
- B. Ludi, M. J. Suess, I. A. Werner and M. Niederberger, Nanoscale, 2012, 4, 1982–1995 RSC.
- D. Li, M. H. Nielsen, J. R. Lee, C. Frandsen, J. F. Banfield and J. J. De Yoreo, Science, 2012, 336, 1014–1018 CrossRef CAS PubMed.
- H. G. Liao, L. Cui, S. Whitelam and H. Zheng, Science, 2012, 336, 1011–1014 CrossRef CAS PubMed.
- Y. Liu, X. M. Lin, Y. Sun and T. Rajh, J. Am. Chem. Soc., 2013, 135(10), 3764–3767 CrossRef CAS PubMed.
- H. Zhang and J. F. Banfield, J. Phys. Chem. Lett., 2012, 3, 2882–2886 CrossRef CAS.
- M. Alimohammadi and K. A. Fichthorn, Nano Lett., 2009, 9, 4198–4203 CrossRef CAS PubMed.
- Y. Qin and K. A. Fichthorn, J. Chem. Phys., 2003, 119, 9745 CrossRef CAS PubMed.
- R. Sathiyanarayanan, M. Alimohammadi, Y. Zhou and K. A. Fichthorn, J. Phys. Chem. C, 2011, 115, 18983–18990 CAS.
- W. D. He, J. H. Lin, X. Lin, N. Lu, M. Zhou and K. H. L. Zhang, Analyst, 2012, 137, 4917–4920 RSC.
- W. D. He, J. H. Lin, B. Wang, S. Q. Tuo, S. T. Pantelides and J. H. Dickerson, Phys. Chem. Chem. Phys., 2012, 14, 4548–4553 RSC.
- W. D. He, CrystEngComm, 2014, 16, 1439–1442 RSC.
- Y. Q. Zhang, W. D. He, J. H. Dickerson, X. Lin, H. L. Lu, K. C. Wen and X. N. Wang, Analyst, 2014, 139, 371–374 RSC.
- W. Q. Lv, W. D. He, X. N. Wang, Y. H. Niu, H. Q. Cao, J. H. Dickerson, Z. G. Wang and R. L. Penn, Nanoscale, 2014, 6, 2531–2547 RSC.
- X. G. Wen, Y. T. Xie, M. W. Cheung, K. Y. Cheung, X. Y. Li, R. Renneberg and S. Yang, Langmuir, 2006, 22, 4836–4842 CrossRef CAS PubMed.
- L. Lu, A. Kobayashi, Y. Kikkawa, K. Tawa and Y. Ozaki, J. Phys. Chem. B, 2006, 110, 23234–23241 CrossRef CAS PubMed.
- B. D. Smith, K. A. Fichthorn and D. J. Kirby, et al., ACS Nano, 2013, 8(1), 657–670 CrossRef PubMed.
- S. A. Harfenist, Z. L. Wang, M. M. Alvarez, I. Vezmar and R. L. Whetten, J. Phys. Chem., 1996, 100, 13904–13910 CrossRef CAS.
- T. K. Sau and C. J. Murphy, Langmuir, 2005, 21, 2923–2929 CrossRef CAS PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- J. E. Lennard-Jones, Proc. R. Soc. London, Ser. A, 1924, 106(738), 463–477 CrossRef.
- B. J. Wiley, Y. Chen, J. McLellan, Y. Xiong, Z. Y. Li, D. Ginger and Y. Xia, Nano Lett., 2007, 7, 1032–1036 CrossRef CAS PubMed.
- R. L. Penn, K. Tanaka and J. Erbs, J. Cryst. Growth, 2007, 309, 97–102 CrossRef CAS PubMed.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Science, 1995, 270, 1335–1338 CAS.
- S. Shen, J. Zhuang, X. Xu, A. Nisar, S. Hu and X. Wang, Inorg. Chem., 2009, 48, 5117–5128 CrossRef CAS PubMed.
- D. Chen and L. Gao, J. Cryst. Growth, 2004, 264(1), 216–222 CrossRef CAS PubMed.
- R. L. Penn and J. F. Banfield, Geochim. Cosmochim. Acta, 1999, 63, 1549 CrossRef CAS.
- X. Xu, J. Zhuang and X. Wang, J. Am. Chem. Soc., 2008, 130, 12527–12535 CrossRef CAS PubMed.
- C. Pacholski, A. Kornowski and H. Weller, Angew. Chem., Int. Ed., 2002, 41, 1188–1191 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra02018b |
| This journal is © The Royal Society of Chemistry 2015 |