
Synthesis and bioactivity of N-glycosylated 3-(2-oxo-2-arylethylidene)-indolin-2-ones†
Dennis
Kleeblatt
a,
Martin
Becker
a,
Michael
Plötz
b,
Madeleine
Schönherr
c,
Alexander
Villinger
a,
Martin
Hein
a,
Jürgen
Eberle
b,
Manfred
Kunz
c,
Qamar
Rahman
ad and
Peter
Langer
*ae
aInstitut für Chemie, Universität Rostock, Albert-Einstein-Str. 3a, 18059 Rostock, Germany. E-mail: peter.langer@uni-rostock.de; Fax: +49 381 4986412
bCharité Centrum 12 für Innere Medizin und Dermatologie, Hauttumorzentrum, Charité – Universitätsmedizin Berlin, Charitéplatz 1, 10117 Berlin, Germany
cKlinik für Dermatologie, Venerologie und Allergologie, Universitätsklinik Leipzig, Philipp-Rosenthal-Str. 23-25, 04103 Leipzig, Germany
dAmity University, Lucknow Campus, Viraj Khand-5, Gomti Nagar, Lucknow-226010, India
eLeibniz-Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany
First published on 16th February 2015
Abstract
N-Glycosyl-3-alkylideneoxindoles, N-glycosylated 3-(2-oxo-2-arylethylidene)indolin-2-ones, were prepared by reaction of isatin-N-glycosides with substituted acetophenones. The biological activities of these new compounds revealed significant antitumor activity in melanoma human cell lines. Inhibition of cell proliferation and loss of cell viability were strongly enhanced by the combination with the death ligand TRAIL (TNF-related apoptosis-inducing ligand). The antitumor effects were related to the inhibition of the survival pathway of c-Jun and JNK2 (Jun N-terminal kinase).
Introduction
The natural product indirubin (1) is an isomer of the blue dye indigo, which has, in contrast to indigo, a violet colour (Fig. 1). During the isolation of indigo from natural sources, as well as during its industrial synthesis, indirubin is formed as a by-product and is responsible for the reddish shine. In addition, indirubin is the pharmacologically active substance of the traditional chinese medicinal recipe Danggui Longhui Wan, which has been used for the treatment of myelocytic leukemia.1 The biological activity of indirubin has been related to the inhibition of cyclin dependent kinases (CDKs).2 During the last years, inhibition of CDKs is becoming an attractive research field for combating cancer and other diseases, which are related to hyperproliferation.3 Indirubin and its substituted derivatives are not only potent inhibitors of cyclin-dependent kinases (CDKs),2 but also of several other kinases, such as glycogen synthase kinase-3 (GSK-3).4 For example, 5-substituted indirubins displayed high inhibitory potency toward various CDKs and GSK-3β.2,4 Also several O- and N-glycosylated indigoid dyes have been synthesized in recent years.5 Many of the compounds studied so far exhibit higher antiproliferative activity toward human cancer cells in vitro than their non-glycosylated analogues. Notably, both deprotected and protected N-glycosides may be of pharmacological relevance.6 For example, the biological activity of so called ‘Natura’, i.e. acetyl-protected β-D-xylopyranosyl-N-isoindigo, was reported to be higher than that of its deprotected analogue.6a | ||
| Fig. 1 Indirubin (1) and 3-alkylideneoxindole 2. | ||
In 2006, our group published the first synthesis of N-glycosylated indirubins.7 We also reported significant activities of several derivatives against various human cancer cell lines. Based on these results, other glycosylated and non-glycosylated derivatives and heteroanalogues of indirubin and isoindigo have been synthesized.8 Some of them, especially the thia-analogous indirubin-N-glycosides, have shown substantial activity against different human cancer cell lines.8c In all these studies, we have found that the presence of a carbohydrate moiety located at the nitrogen atom of the heterocycle resulted in a significant increase of the anti-cancer activity.
In the course of our program related to the development of new, biologically active indirubin derivatives, we turned our attention to 3-alkylideneoxindoles, such as 2, which can be regarded as structurally simplified analogues of indirubin (Fig. 1). We assumed that these structurally more flexible structures might better interact with the reactive center of an enzyme, which should lead to an enhanced biological activity. Woodard et al. studied the activity of several non-glycosylated 3-alkylideneoxindoles as CDK inhibitors.9a They reported a very high inhibition in case of derivative 3b containing a halogen substituent. In contrast, only a weak inhibition was observed for unsubstituted derivative 3a (Fig. 2). Belenkaya et al. reported anti-malaria activity of nitro substituted compound 3f.9b
 | ||
| Fig. 2 Known biologically active non-glycosylated 3-alkylideneoxindoles. | ||
Herein, we report what is, to the best of our knowledge, the first synthesis of N-glycosylated 3-alkylideneoxindoles and their antitumor activity in human melanoma cell lines. During the optimization of the biological activity, the most promising substitution patterns were selected based on known non-glycosylated derivatives.9
Results and discussion
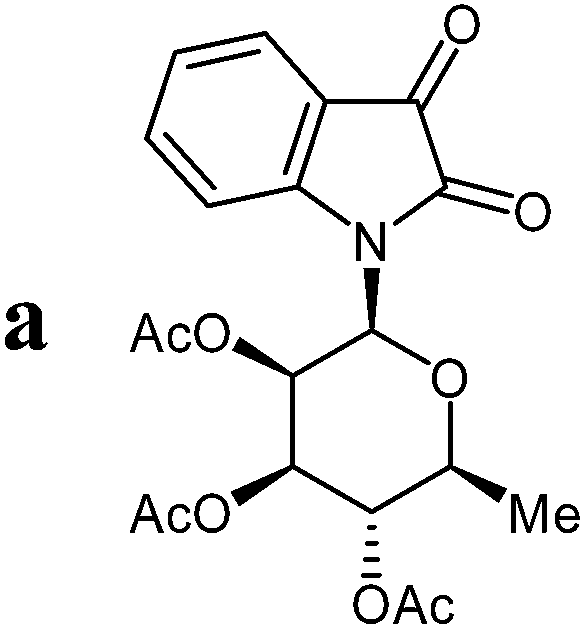
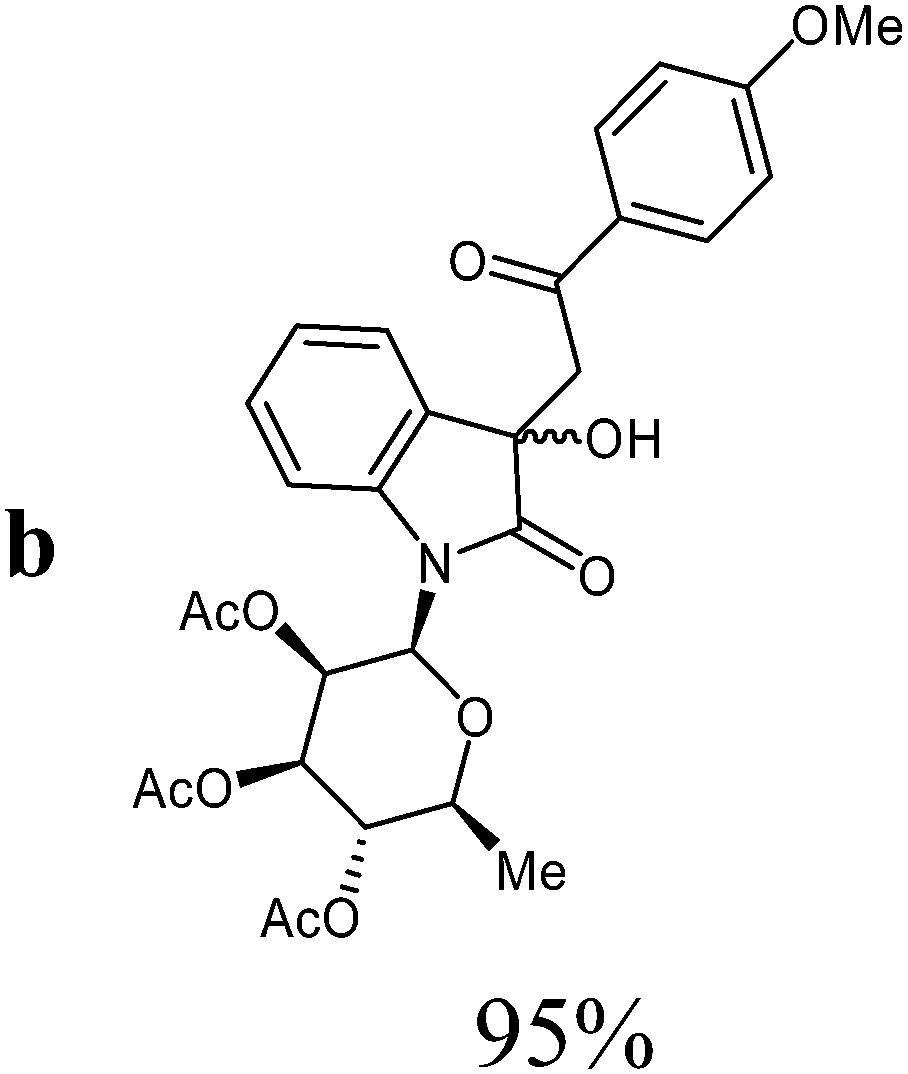
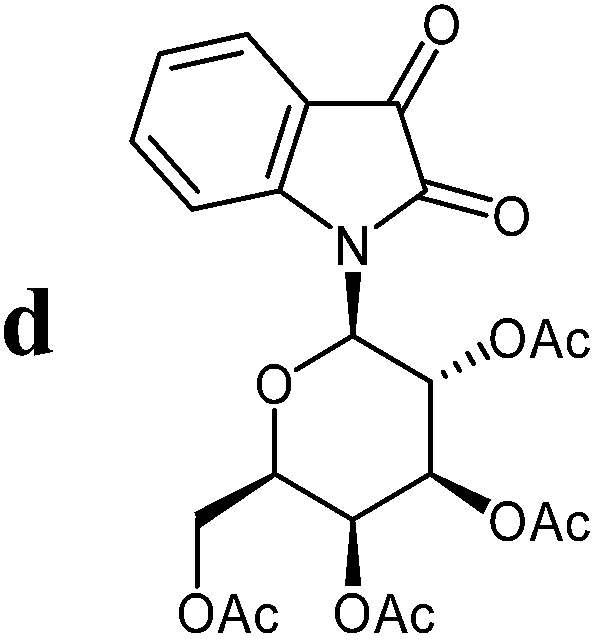
Isatin-N-glycoside 7a was prepared, following our previously reported procedure,8 by reaction of L-rhamnose with aniline, acetylation and subsequent cyclization with oxalyl chloride (Scheme 1). Likewise, derivatives 7b, 7c and 7d were prepared from D-mannose, D-glucose and D-galactose, respectively.6b,7b,10 The condensation of isatin-N-glycoside 7a with acetophenone afforded N-glycosylated 3-alkylideneoxindole 9a in up to 71% yield (Scheme 2). The reaction was optimized based on our previous experience with indirubin-N-glycosides and their heteroanalogous compounds,8 and based on known chemistry of non-glycosylated 3-alkylideneoxindoles.9 We tried several conditions for the condensation reaction and found that a two-step procedure worked best in our hands. The first step was the aldol reaction of the acetophenone with the carbonyl group (C-3) of the isatin-N-glycoside (Scheme 2). For this reaction step we used triethylamine as the base and ethanol as the solvent at a temperature of 20 °C. Only in case of parent acetophenone no additional solvent was used. In two cases we could isolate and characterize the aldol addition products, which were formed as diastereomeric mixtures, because of the new stereocenter at C-3. The ratio of the diastereomers was found to be about 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, but we did not investigate the configuration of the main/minor isomer at C-3. In general, it was sufficient to isolate the crude products, by simple distillation of the solvent from the reaction mixture, to be used as the starting materials in the following step. The purified or the crude products were reacted with mesyl chloride, in the presence of triethylamine (and in some cases in the additional presence of DMAP), to give the desired dehydrated final products. After workup and purification, the products could be isolated in 71% yield.
:1, but we did not investigate the configuration of the main/minor isomer at C-3. In general, it was sufficient to isolate the crude products, by simple distillation of the solvent from the reaction mixture, to be used as the starting materials in the following step. The purified or the crude products were reacted with mesyl chloride, in the presence of triethylamine (and in some cases in the additional presence of DMAP), to give the desired dehydrated final products. After workup and purification, the products could be isolated in 71% yield.
 | ||
| Scheme 1 Synthesis of 1-(2,3,4-tri-O-acetyl-β-L-rhamnopyranosyl)isatin (7a). Reagents and conditions: (i) PhNH2, EtOH (abs.), 20 °C, 24 h; (ii) Ac2O, pyridine, 0 °C, 12 h; (iii) oxalyl chloride (10 equiv.), AlCl3 (1 equiv.), 55 °C, 1.5 h, then chromatographic separation of the anomers. | ||
 | ||
| Scheme 2 Synthesis of 9a. Reagents and conditions: (i) NEt3 (1 eq.), EtOH 20 °C, 6–10 h. (ii) MsCl, NEt3, (DMAP), CH2Cl2, 0–20 °C, 2 h, 20 °C, 6–8 h. | ||
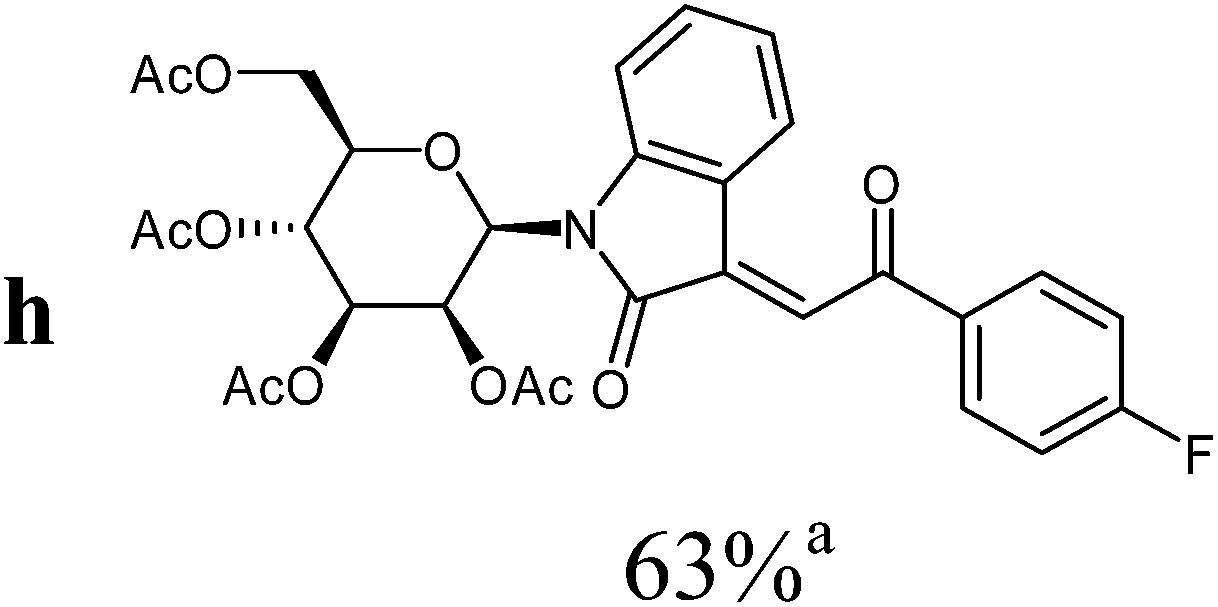
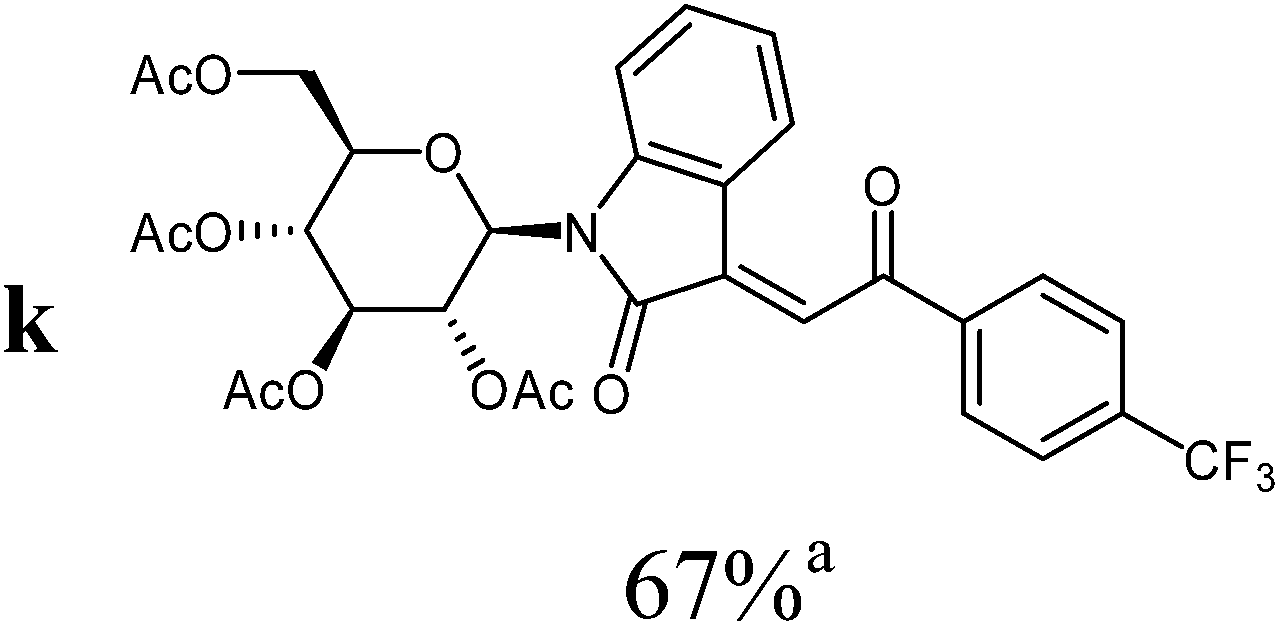
The preparative scope was next studied (Table 1). The condensation of isatin-N-glycoside 7a–d with various substituted acetophenones afforded N-glycosyl-3-alkylideneoxindoles 9a–m in 45–78% yields (based on 7).
| 7 | 8 yield | 9 yield | IC50 (µM) (mean ± SD) |
|---|---|---|---|
| a Yields of isolated products 9 based on 7 using crude intermediate 8 for the dehydratisation step. b Yields of isolated products 9 based on purified intermediate 8 used for the dehydratisation step. | |||

|

|

|
— |
|
|

|
4.4 ± 0.5 |

|
|
4.3 ± 0.7 | |

|
|
2.8 ± 0.6 | |

|

|
4.2 ± 0.4 | |

|

|
6.0 ± 0.6 | |

|

|
— | |

|
|
7.0 ± 0.7 | |

|

|
5.9 ± 0.5 | |

|

|
— | |

|
|
— | |

|

|
— | |
|

|
— | |
For the overall reaction, using crude intermediates 8 for the final dehydratisation step, we observed a trend to higher yields for acetophenones containing electron-withdrawing substituents. The highest yields were observed for the nitro-substituted derivative, while the lowest yield was observed for the methoxy-substituted derivatives. This result is surprising, because of the lower nucleophilicity of the acceptor-substituted acetophenones.
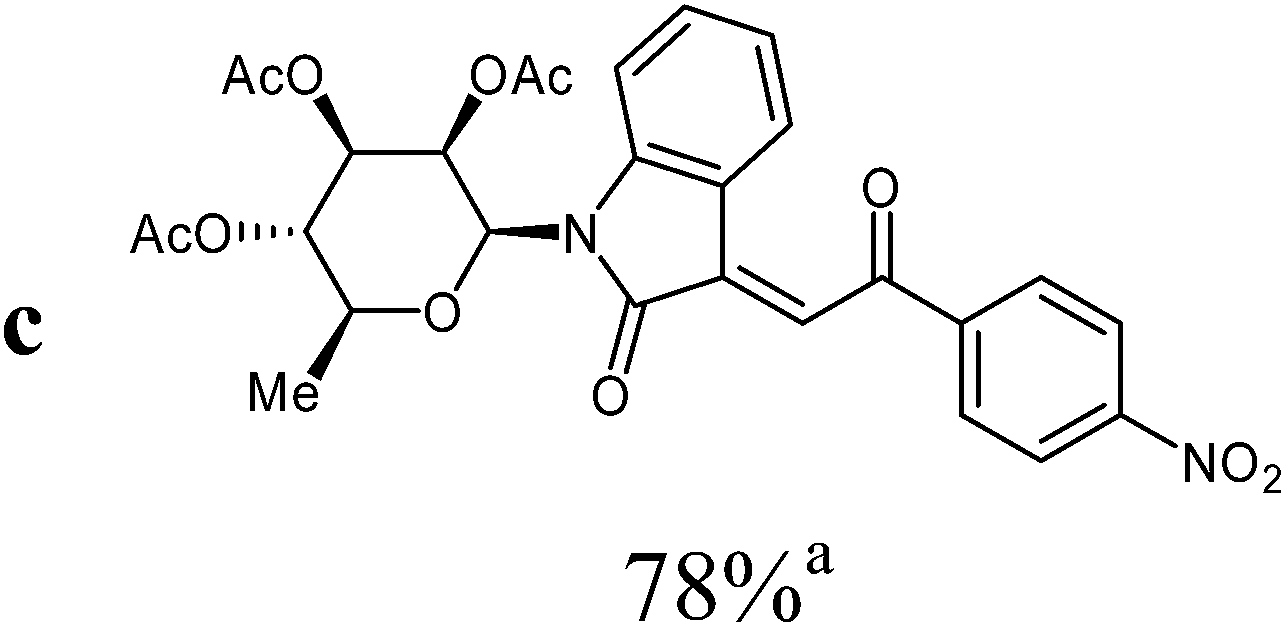
For the structure elucidation of compounds 9 two dimensional NMR experiments (1H,1H-COSY, HSQC and 1H,1H-NOESY) were carried out. In the 1H,1H-NOESY spectra, the missing correlation between H-8 and H-4 (Fig. 3) was a first, but unreliable indication of the E-configuration of the double bond between C-3 and C-8. Furthermore, the β-configuration of the carbohydrate substituents could be clearly shown by the correlation signals of H-1′, H-3′ and H-5′ in the 1H,1H-NOESY spectra (only β-configured starting isatin-N-glycosides were applied for the reactions). These correlations can appear in case of β-L-rhamno configured substituents, when they have a 1C4- (or similar) conformation, as well as in case of the other investigated carbohydrate substituents, when they have a 4C1-conformation. The above mentioned correlations were observed for all studied examples. Additionally, the structure of 9c was independently confirmed by X-ray crystal structure analysis (Fig. 4).11 Here, the E-configuration of the double bond as well as the anomeric β-configuration and 1C4-conformation of the sugar moiety was clearly proven. Because of comparable NMR data of all the 3-(2-oxo-2-arylethylidene)indolin-2-ones, we assumed that all these compounds possess an E-configured double bond.
 | ||
| Fig. 3 1H,1H-NOESY correlations for compound 9c. | ||
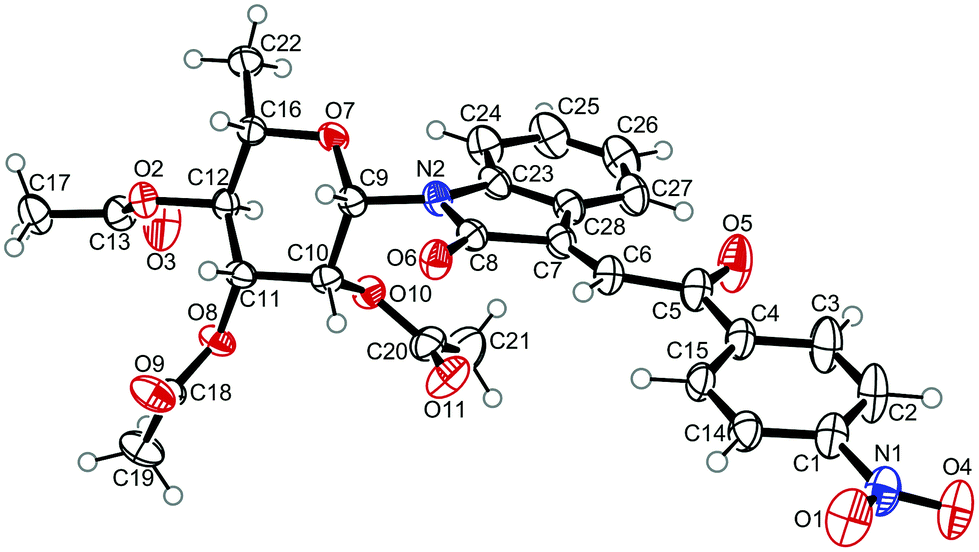 | ||
| Fig. 4 X-ray structure image of compound 9c (ORTEP plot with displacement ellipsoids at the 50% probability level). | ||
We also tried to remove the acetyl protecting groups from the carbohydrate substituents of compounds 9. Therefore, typical methods under basic (e.g. 1% methanolic sodium methanolate)12 as well as acidic conditions (hydrogen chloride in methanol) were applied. However, it was not possible to isolate the deprotected products from the reaction mixture. The result can be explained by a retro-aldol reaction as the formation of the corresponding acetophenone and of the isatin-N-glycoside was detected. However, as mentioned in the introduction, acetyl-protected glycosylated isoindigo and indirubin derivatives proved, in some cases even more active than their deprotected analogues.6a Therefore, we studied the pharmacological activity of our acetylated products and, in fact, interesting biological effects were observed which are outlined below.
In previous studies, we had investigated the biological activity of compound 9b in melanoma cell lines SK-Mel-19, SK-Mel-29, SK-Mel-103 and SK-Mel-147 and observed only moderate direct activities in these cells.8c In the present work, we studied the growth inhibitory activities (IC50) of selected examples of various derivatives of derivatives 9 (Table 1). Here, we focussed on cell line SK-Mel-147 in order to put an emphasis on the differences between the individual substances. This cell line is characterized by a very high proliferative activity, which may mirror the situation of highly aggressive metastatic cells in the human system. We selected the derivatives for biotests only those compounds which are stable in water for an extended period of time. Compounds which tend to be slightly unstable in aqueous solution were not tested in order to avoid misleading biological results. Those derivatives containing a β-L-rhamnosyl substituent revealed slightly higher activities than the mannosylated compounds. The highest activity was found for the chloro-substituted derivative 9d. The higher growth inhibitory activity of derivative 9b in our current experiments (IC50 value: 4.4 ± 0.5), as compared to our earlier results (IC50 value: 27.2 ± 1.3),8c may be explained by (i) culture conditions, (ii) the different incubation times applied (48 h in current vs. 24 h in previous experiments) as well as by (iii) two different assays, used (WST-1 in previous vs. XTT assay in current experiments). In addition, human benign dermal fibroblasts were used here as controls, and the IC50 values of indirubin derivatives were significantly higher in fibroblasts than in melanoma cells (9b: 8.8 ± 1.1 µM; 9c: 32.9 ± 0.9 µM; 9d: 9.1 ± 0.7 µM; 9e: 9.5 ± 0.3 µM; 9f: 11 ±1.5 µM; 9h: 19.3 ± 0.9 µM; 9i: 9.6 ±2.4 µM; cf. Table 1).
Further biological effects of the N-glycosylated 3-(2-oxo-2-arylethylidene)indolin-2-one derivatives 9c–f, 9i–k and 9m were exemplary investigated in the frequently investigated human melanoma cell line A-375,13 which is well characterized for apoptosis regulation. In particular, apoptosis regulation by the cytokine and death ligand TRAIL (TNF-related apoptosis-inducing ligand) is well described in this cell line.14,15 Cell viability was monitored by calcein staining and flow cytometry. The applied concentrations of 10 µM of the indirubin derivatives resulted in a significant reduction of cell viability after 24 h (20–30%; Fig. 5a). We had previously reported that indirubin derivatives, as also their heteroanalogous compounds, can strongly enhance apoptosis induced by death ligand TRAIL.8,14 Thus, combinations with TRAIL were also tested for the new series of N-glycosylated 3-(2-oxo-2-arylethylidene)indolin-2-ones. Whereas TRAIL (20 ng mL−1) alone showed only moderate effects on A-375 cell viability (20% reduction), the combinations dramatically reduced the numbers of viable cells (50–90% reduction; Fig. 5a).
 | ||
| Fig. 5 Effects of N-glycosylated 3-(2-oxo-2-arylethylidene)indolin-2-ones on melanoma cells: (a) cell viability of A-375 cells was determined after 24 h by calcein staining. Cells were treated with TRAIL (20 ng mL−1), compounds of the series 9 (10 µM) or with combinations. (b) Cell proliferation of A-375 cells expressed as percent of non-treated controls was determined after 48 h by WST-1 assay. Cells were treated with 1.25 µM of compounds of series 9 and/or 20 ng mL−1 TRAIL. (c) Real-time cell analysis (RTCA) of A-375 cells treated with 1.25 µM/2.5 µM of 9c–f, 9i, 9m, 9j and 9k ± 20 ng mL−1 TRAIL. Seeding density was 1250 cells per microtiter well. The cell index was normalized at 24 h, when treatment was started. Cell indices indicate cell adhesion. (a–c) Each two independent experiments with triplicate determinations revealed highly comparable results. | ||
Decreased cell viability also exerted a strong impact on cell proliferation, monitored in A-375 cells here by WST-1 assay. Thus, inhibition of cell proliferation upon combination treatment with TRAIL ranged between 70% and 90% (Fig. 5b). Direct cytotoxicity on the other hand remained unaffected at this time, as determined by LDH release in cell culture supernatants (data not shown). Cell proliferation was also determined in real time. In fact, A-375 cells were still proliferative when treated with TRAIL (20 ng mL−1) or several indirubin derivatives alone, e.g. 9c, 9f at 1.25 µM and 9e, 9i, 9m, 9k at 2.5 µM. Inhibition by these indirubin derivatives alone ranged between 40% and 80% at a time point of 100 h (75 h after treatment). At this time, inhibition by TRAIL alone was at 60%. However upon combination treatments, cell proliferation and cell attachment was almost completely abolished for all tested indirubins. For 9d and 9j, an enhanced effect by combination treatment could not be clearly shown, as in this assay the effect of 9d and 9j alone was already very strong (Fig. 5c).
As reported previously, treatment of melanoma cells with glycosylated indirubin analoga resulted in an inhibition of the phosphorylation of the transcription factor c-Jun.8c This may be due to a direct inhibition of its upstream kinases c-Jun N-terminal kinases 1/2 (JNK1/2). To further address this question, SK-Mel-147 cells were treated with derivative 9b and c-Jun phosphorylation was determined by immunoblotting. As treatment with 10 µM of 9b was associated with significant induction of cell death, lower concentrations were used (5 µM, 6.5 µM and 7.5 µM). Inhibited c-Jun phosphorylation was seen for all three concentrations, and strongest effects were obtained with 6.5 µM (Fig. 6). The three concentrations also induced upregulation of p53 levels, which may support apoptosis induction in melanoma cells via p53-dependent pathways.
 | ||
| Fig. 6 Derivative 9b suppresses c-Jun phosphorylation in melanoma cells. SK-Mel-147 melanoma cells were treated for indicated times with 6.5 µM of 9b. Total protein lysates of cells were subjected to immunoblotting using specific antibodies for phosphorylated c-Jun (Ser63/73) and total p53. Staining for GAPDH was used as loading control. | ||
To test whether inhibition of c-Jun phosphorylation was due to a direct inhibitory effect on the upstream kinases JNK1/2, ATP-competitive kinase binding assays were performed in vitro using recombinant JNK2 kinase. The small molecule inhibitor sorafenib, a well-known multi-kinase inhibitor which strongly inhibits JNK1/2 kinases, was used as positive control. The IC50 of sorafenib for the inhibition of JNK2 was determined at 1.6 µM, whereas the IC50 of 9b was at 1.06 µM. As negative control, the indirubin derivative 10 (Fig. 7)7 was used, which did not inhibit the activity of JNK2 (Fig. 8).
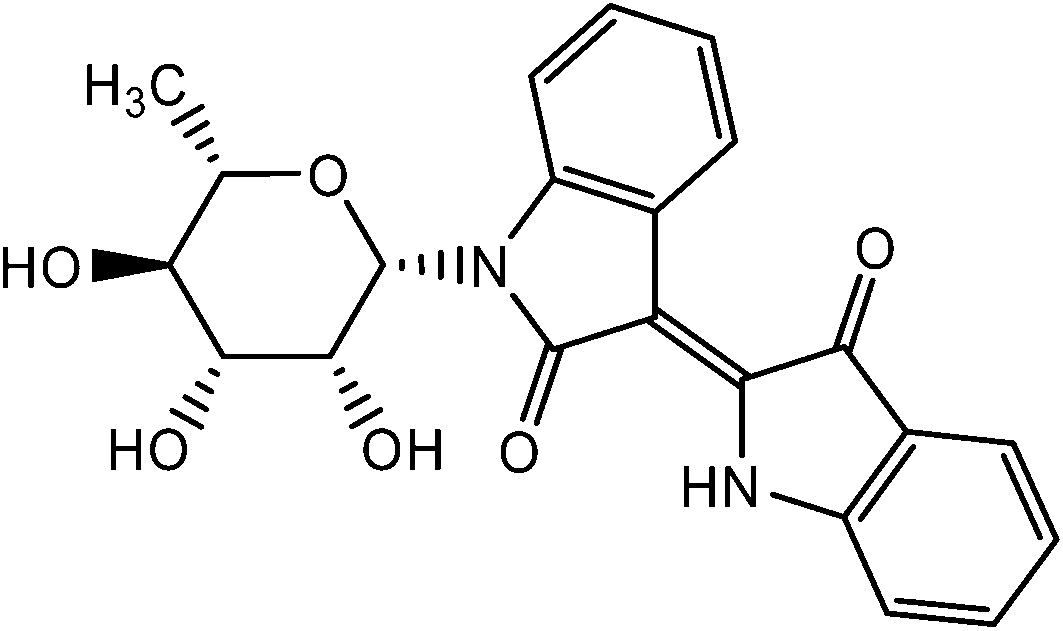 | ||
| Fig. 7 Structure of the N-β-L-rhamnopyranosylindirubin 10 used as control substance. | ||
 | ||
| Fig. 8 Derivative 9b strongly inhibits JNK2 activity in melanoma cells. Kinase ATP-binding assays were performed with recombinant JNK2 using different concentrations of the positive control inhibitor sorafenib as well as of derivatives 9b and 10. Experiments were performed three times each in triplicates. IC50 values were calculated after analysis of all experiments. Graphs correspond to the means of one representative experiment. | ||
Thus, it could be demonstrated that derivative 9b exerted a strong inhibitory effect on JNK2, which explains its inhibitory activity on c-Jun phosphorylation. Suppression of JNK2 and c-Jun may significantly contribute to the decreased proliferation and enhanced apoptosis sensitivity of melanoma cells after treatment with derivative 9b, as c-Jun plays a decisive role in melanoma cell proliferation and survival.
Conclusions
In conclusion, the first synthesis of acetyl protected N-glycosylated 3-(2-oxo-2-arylethylidene)indolin-2-ones was reported. The synthesis relies on the aldol condensation of readily available isatin-N-glycosides with substituted acetophenones. The preparation of the corresponding deprotected derivatives failed in most of the cases, probably due to the preferred formation of products of the retroaldol reaction. However, the protected derivatives showed interesting pharmacological activities, such as a significant decrease of cell viability and inhibition of cell proliferation. The effects were stronger in combination with the proapoptotic death ligand TRAIL (TNF-related apoptosis-inducing ligand). A reduction of the cell viability by up to 90% and complete abrogation of cell proliferation are strongly suggestive for testing therapeutic strategies. With regard to our investigations related to the mode of antitumor activity of indirubin derivatives in the cell, our present study suggests that the investigated derivatives affected the prosurvival pathway of JNK and c-Jun.Experimental section
Material and methods for the syntheses and characterisation of the new compounds
All solvents were anhydrous and commercially available and all reactions were performed under argon atmosphere. The synthesis of N-glycosylated isatin derivatives has been described in the literature.6b,7b,10 The substituted acetophenones were commercially available. Yields refer to isolated products. 1H NMR spectra (250.13 MHz, 300.13 MHz and 500.13 MHz) and 13C NMR spectra (62.9 MHz, 75.5 MHz and 125.8 MHz) were recorded on Bruker spectrometers AV 250, AV 300 and AV 500 in CDCl3 and DMSO-d6 as solvents. The calibration of spectra was carried out on solvent signals (CDCl3: δ (1H) = 7.25, δ (13C) = 77.0); acetone-d6: δ (1H) = 2.05, δ (13C) = 29.83). For the NMR signals of the compounds 8a, b and 9a–m the atom labelling given in Fig. 3 was used. Mass spectrometric data (MS) were obtained by electron ionization (EI, 70 eV), chemical ionization (CI, isobutane) or electrospray ionization (ESI). The mass spectra and high-resolution mass spectra (HRMS) were recorded on the following MS instruments: Time-of-Flight (LC/MS 6210 (Agilent Technologies) for ESI and Finnigan MAT 95 XP (Thermo Electron Corporation) for EI/CI. Elemental analyses were performed on a C,H,N,S analyser (Thermo Quest Flash EA 1112). The melting points were measured with a polarizing microscope (Leitz Laborlux 12 Pol-S) equipped with a hot stage (Mettler FP 90). Thin layer chromatography was performed on silica-gel foil (Merck DC-foil, silica gel 60, F254) and silica gel plates (Merck HPTLC precoated plates, silica gel 60, F254) pursues. Detection was carried out via UV absorbance at 254 nm or 366 nm and/or development with 10% methanolic sulfuric acid and subsequent heat treatment. Column chromatography was performed on Macherey-Nagel silica gel 60 (particle size 63–200 nm, 70–230 mesh).General procedure for the synthesis of the intermediate addition products 8 and aldolcondensation products 9
1 eq. of the N-glycosylisatin and 2 eq. of the substituted acetophenone are dissolved in abs. ethanol (4–8 mL per mmol N-glycosylisatin) (in the case of the unsubstituted acetophenone as reactant, this was used as the solvent in excess and no ethanol was used). To this solution, 1–2 eq. of triethylamine are added and the mixture is stirred for 6–12 hours at room temperature. After complete conversion of the starting material (TLC control), the reaction mixture is diluted with toluene and then evaporated under reduced pressure. The crude aldol addition products 8 can be directly used for the next step or (if containing larger amounts of by-products) can be purified by column chromatography (heptane:ethyl acetate, v:v = 3:1 → 2:1).
The crude or purified aldol addition product 8 (1 eq.) and 4-(dimethylamino)pyridine (1.1 eq.)* are dissolved in dry dichloromethane and the solution is cooled to 0 °C. Then triethylamine (1.1 eq.) (*in the cases were no DMAP was used, 2.2 eq. of NEt3 were added) and methanesulfonyl chloride (1.1 eq.) are carefully added (syringe). After that, the cooling bath is removed and the mixture is allowed to warm to room temperature during 2 hours. After additional 6–8 hours stirring at room temperature (TLC control), the solution is diluted with dichloromethane and washed twice with saturated NaHCO3 solution, 1 M NaHSO4 solution and brine, respectively. The organic layer is then dried over sodium sulfate, filtered and concentrated under diminished pressure. The residue is purified by column chromatography (heptane:ethyl acetate, v:v = 2:1) to give product 9.
:ethyl acetate v:v = 6:1 → 3:1) as colourless amorphous solid (yield: 238 mg, 93%) which consists of a diastereomeric mixture (diasteromer 1:diasteromer 2 = 4:1) due to the new formed chiral center at C-3. Rf 0.15 (heptane:ethyl acetate, v:v = 3:1).
1H NMR (250 MHz, CDCl3): diasteromer 1: δ = 7.87 (d, 3Jo,m = 7.9 Hz, 2H, 2Hortho), 7.54 (“t”, 3Jm,p = 7.6 Hz, 1H, Hpara), 7.53 (d, 3J7,6 = 7.5 Hz, 1H, H-7), 7.41 (“t”, 3Jo,m ≈ 3Jm,p ≈ 7.8 Hz, 2H, 2Hmeta), 7.32 (d, 3J4,5 = 7.5 Hz, 1H, H-4), 7.23 (“t”, 3J6,7 ≈ 3J6,5 ≈ 7.6 Hz, 1H, H-6), 6.98 (“t”, 3J5,4 ≈ 3J5,6 ≈ 7.6 Hz, 1H, H-5), 5.85 (d, 3J1′,2′ = 1.5 Hz, H-1′), 5.59 (dd, 3J2′,1′ = 1.5 Hz, 3J2′,3′ = 3.0 Hz, 1H, H-2′), 5.26–5.21 (m, 2H, H-3′, H-4′), 3.79 (s, 1H, OH), 3.78–3.66 (m, 1H, H-5′), 3.71 (d, 2J8a,8b = 17.2 Hz, 1H, H-8a), 3.42 (d, 2J8a,8b = 17.2 Hz, 1H, H-8b), 2.08, 1.98, 1.77 (3s, 9H, 3CH3CO), 1.35 (d, 3J5′,6′ = 6.2 Hz, 3H, 3H-6′). Diasteromer 2 (minor): δ = 7.87 (d, 3Jo,m = 7.9 Hz, 2H, 2Hortho), 7.54 (“t”, 3Jm,p = 7.6 Hz, 1H, Hpara), 7.53 (d, 3J7,6 = 7.5 Hz, 1H, H-7), 7.41 (“t”, 3Jo,m ≈ 3Jm,p ≈ 7.8 Hz, 2H, 2Hmeta), 7.32 (d, 3J4,5 = 7.5 Hz, 1H, H-4), 7.23 (“t”, 3J6,7 ≈ 3J6,5 ≈ 7.6 Hz, 1H, H-6), 6.98 (“t”, 3J5,4 ≈ 3J5,6 ≈ 7.6 Hz, 1H, H-5), 5.87 (d, 3J1′,2′ = 1.4 Hz, H-1′), 5.59 (dd, 3J1′,2′ = 1.4 Hz, 3J2′,3′ = 3.0 Hz, 1H, H-2′), 5.26–5.21 (m, 2H, H-3′, H-4′), 4.12 (d, 2J8a,8b = 14.1 Hz, 1H, H-8a), 4.08 (d, 2J8a,8b = 14.1 Hz, 1H, H-8b), 3.79 (s, 1H, OH), 3.78–3.66 (m, 1H, H-5′), 2.08, 1.98, 1.68 (3s, 9H, 3CH3CO), 1.40 (d, 3J5′,6′ = 6.2 Hz, 3H, 3H-6′). 13C NMR (75 MHz, CDCl3): diasteromer 1: δ = 198.5 (C-9), 175.3 (C-2), 170.1, 170.0, 169.7 (3CH3CO), 141.0 (C-7a), 136.3 (Cipso), 133.8 (Cpara), 129.9 (C-3a), 129.2 (C-6), 128.7 (2Cmeta), 128.3 (2Cortho), 123.5 (C-4), 123.1 (C-5), 114.2 (C-7), 80.7 (C-1′), 74.3 (C-3), 73.9 (C-5′), 70.5 (C-2′), 70.4 (C-3′), 70.3 (C-4′), 44.2 (C-8), 20.9, 20.8, 20.6 (3CH3CO), 17.7 (C-6′). Diasteromer 2 (minor): δ = 196.8 (C-9), 175.7 (C-2), 171.1, 170.0, 169.6 (3CH3CO), 141.5 (C-7a), 136.1 (Cipso), 133.6 (Cpara), 130.1 (C-3a), 129.4 (C-6), 128.6 (2Cmeta), 128.1 (2Cortho), 123.3 (C-4), 123.1 (C-5), 114.0 (C-7), 81.2 (C-1′), 74.0 (C-3), 73.6 (C-5′), 70.4 (C-2′), 70.3 (C-3′), 70.1 (C-4′), 44.8 (C-8), 20.9, 20.8, 20.6 (3CH3CO), 17.7 (C-6′). HRMS (ESI): calc. for C28H30NO10 ([M + H]+) 540.18642, found 540.18695.
:ethyl acetate v:v = 6:1 → 3:1) as colourless amorphous solid (yield: 260 mg, 95%) which consists of a diastereomeric mixture (diasteromer 1:diasteromer 2 = 3:1) due to the new formed chiral center at C-3. Rf 0.22 (heptane:ethyl acetate, v:v = 3:1).
1H-NMR (250 MHz, CDCl3): diasteromer 1: δ = 7.91–7.83 (m, 3Jo,m = 7.9 Hz, 2H, 2Hortho), 7.50 (d, 3J7,6 = 7.9 Hz, 1H, H-7), 7.32 (d, 3J4,5 = 7.6 Hz, 1H, H-4), 7.22 (“dt”, 4J6,4 = 1.2 Hz, 3J6,7 ≈ 3J6,5 ≈ 7.8 Hz, 1H, H-6), 6.98 (“dt”, 4J5,7 = 1.3 Hz, 3J5,4 ≈ 3J5,6 ≈ 7.6 Hz, 1H, H-5), 6.88 (“d”, 3Jo,m = 7.8 Hz, 2H, 2Hmeta), 5.83 (d, 3J1′,2′ = 1.4 Hz, H-1′), 5.58 (dd, 3J1′,2′ = 1.5 Hz, 3J2′,3′ = 3.0 Hz, 1H, H-2′), 5.25–5.20 (m, 2H, H-3′, H-4′), 5.00 (s, 1H, OH), 3.83 (s, 3H, OCH3), 3.77–3.69 (m, 1H, H-5′), 3.59 (d, 2J8a,8b = 16.8 Hz, 1H, H-8a), 3.29 (d, 2J8a,8b = 16.8 Hz, 1H, H-8b), 2.08, 1.98, 1.76 (3s, 9H, 3CH3CO), 1.35 (d, 3J5′,6′ = 6.2 Hz, 3H, 3H-6′). Diasteromer 2 (minor): δ = 7.91–7.83 (m, 3Jo,m = 7.9 Hz, 2H, 2Hortho), 7.50 (d, 3J7,6 = 8.0 Hz, 1H, H-7), 7.32 (d, 3J4,5 = 7.6 Hz, 1H, H-4), 7.22 (“dt”, 4J6,4 = 1.2 Hz, 3J6,7 ≈ 3J6,5 ≈ 7.8 Hz, 1H, H-6), 6.98 (“dt”, 4J5,7 = 1.3 Hz, 3J5,4 ≈ 3J5,6 ≈ 7.6 Hz, 1H, H-5), 6.88 (“d”, 3Jo,m = 7.8 Hz, 2H, 2Hmeta), 5.86 (d, 3J1′,2′ = 1.4 Hz, H-1′), 5.58 (dd, 3J1′,2′ = 1.5 Hz, 3J2′,3′ = 3.0 Hz, 1H, H-2′), 5.25–5.20 (m, 2H, H-3′, H-4′), 4.12 (d, 2J8a,8b = 14.4 Hz, 1H, H-8a), 4.09 (d, 2J8a,8b = 14.4 Hz, 1H, H-8b), 4.03 (s, 1H, OH), 3.83 (s, 3H, OCH3), 3.77–3.69 (m, 1H, H-5′), 2.08, 1.97, 1.69 (3s, 9H, 3CH3CO), 1.39 (d, 3J5′,6′ = 6.2 Hz, 3H, 3H-6′). 13C-NMR (75 MHz, CDCl3): diasteromer 1: δ = 197.4 (C-9), 175.3 (C-2), 170.2, 170.0, 169.6 (3CH3CO), 164.1 (Cpara), 140.8 (C-7a), 130.8 (2Cmeta), 130.1 (Cipso), 129.5 (C-3a), 129.1 (C-6), 123.6 (C-4), 123.1 (C-5), 113.9 (2Cortho), 113.8 (C-7), 80.6 (C-1′), 74.5 (C-3), 73.9 (C-5′), 70.5 (C-2′), 70.4 (C-3′), 70.3 (C-4′), 55.5 (OCH3), 43.2 (C-8), 20.8, 20.7, 20.5 (3CH3CO), 17.7 (C-6′). Diasteromer 2 (minor): δ = 195.5 (C-9), 175.7 (C-2), 170.0, 170.0, 169.6 (3CH3CO), 163.9 (Cpara), 141.4 (C-7a), 130.6 (2Cmeta), 130.3 (Cipso), 129.3 (C-3a), 129.3 (C-6), 123.3 (C-4), 123.1 (C-5), 113.8 (2Cortho), 113.6 (C-7), 81.1 (C-1′), 73.9 (C-5′), 73.8 (C-3), 70.4 (C-2′), 70.4 (C-3′), 70.1 (C-4′), 55.5 (OCH3), 44.2 (C-8), 20.7, 20.6, 20.5 (3CH3CO), 17.7 (C-6′). MS (EI, 70 eV): m/z (%) = 569 (M+, 1), 551 (4), 273 (45), 171 (15), 153 (53). HRMS (ESI): calc. for C29H32NO11 ([M + H]+) 570.19699, found 570.19621.
:ethyl acetate v:v = 6:1 → 3:1) as orange solid (yield: 137 mg, 71%). mp 74–76 °C (heptane:ethyl acetate).
1H-NMR (300 MHz, CDCl3): δ = 8.22 (d, 3J4,5 = 7.9 Hz, 1H, H-4), 8.08–8.05 (m, 2H, 2Hortho), 7.83 (s, 1H, H-8), 7.61 (“t”, 3J = 7.8 Hz, 2H, 2Hmeta), 7.53–7.47 (m, 1H, Hpara), 7.48 (d, 3J7,6 = 7.6 Hz, 1H, H-7), 7.30 (“dt”, 4J6,4 = 1.2 Hz, 3J6,7 ≈ 3J6,5 ≈ 7.6 Hz, 1H, H-6), 6.98 (“dt”, 4J5,7 = 1.2 Hz, 3J5,4 ≈ 3J5,6 ≈ 7.8 Hz, 1H, H-5), 5.94 (d, 3J1′,2′ = 1.4 Hz, H-1′), 5.59 (dd, 3J1′,2′ = 1.5 Hz, 3J2′,3′ = 3.0 Hz, 1H, H-2′), 5.29–5.17 (m, 3J2′,3′ = 3.0 Hz, 3J3′,4′ = 10.1 Hz, 2H, H-3′, H-4′), 3.80–3.71 (m, 3J5′,6′ = 6.1 Hz, 3J4′,5′ = 9.4 Hz, 1H, H-5′), 2.09, 1.97, 1.84 (3s, 9H, 3CH3CO), 1.35 (d, 3J5′,6′ = 6.2 Hz, 3H, 3H-6′). 13C-NMR (75 MHz, CDCl3): δ = 191.1 (C-9), 169.9, 169.7, 169.6 (3CH3CO), 166.9 (C-2), 143.2 (C-3), 137.4 (Cipso), 135.2 (C-7a), 133.9 (Cpara), 131.9 (C-6), 128.9 (2Cmeta), 128.8 (2Cortho), 127.2 (C-8), 127.1 (C-4), 122.9 (C-5), 120.2 (C-3a), 113.9 (C-7), 80.4 (C-1′), 74.0 (C-5′), 70.6 (C-3′), 70.4 (C-2′), 70.1 (C-4′), 20.8, 20.7, 20.5 (3CH3CO), 17.6 (C-6′). MS (EI, 70 eV): m/z (%) = 521 (M+, 12), 273 (24), 153 (59). HRMS (ESI): calc. for C28H27NNaO9 ([M + Na]+) 544.15780, found 544.15760.
:ethyl acetate v:v = 6:1 → 3:1) as orange solid (yield: 163 mg, 80%). Mp 87–90 °C (heptane:ethyl acetate). Rf 0.45 (heptane:ethyl acetate, v:v = 1:3).
1H-NMR (300 MHz, CDCl3): δ = 8.14 (d, 3J4,5 = 7.9 Hz, 1H, H-4), 8.06 (d, 3Jo,m = 8.9 Hz, 2H, 2Hortho), 7.83 (s, 1H, H-8), 6.97 (d, 3Jo,m = 8.9 Hz, 2H, 2Hmeta), 7.50 (d, 3J7,6 = 7.9 Hz, 1H, H-7), 7.30 (“dt”, 4J6,4 = 1.2 Hz, 3J6,7 ≈ 3J6,5 ≈ 7.7 Hz, 1H, H-6), 6.98 (“dt”, 4J5,7 = 1.2 Hz, 3J5,4 ≈ 3J5,6 ≈ 7.8 Hz, 1H, H-5), 5.94 (d, 3J1′,2′ = 1.4 Hz, 1H, H-1′), 5.59 (dd, 3J1′,2′ = 1.5 Hz, 3J2′,3′ = 3.0 Hz, 1H, H-2′), 5.29–5.17 (m, 3J2′,3′ = 3.0 Hz, 3J3′,4′ = 10.1 Hz, 2H, H-3′, H-4′), 3.88 (s, 3H, OCH3), 3.80–3.71 (m, 3J5′,6′ = 6.1 Hz, 3J4′,5′ = 9.4 Hz, 1H, H-5′), 2.09, 1.97, 1.84 (3s, 9H, 3CH3CO), 1.35 (d, 3J5′,6′ = 6.2 Hz, 3H, 3H-6′). 13C-NMR (75 MHz, CDCl3): δ = 189.6 (C-9), 169.9, 169.7, 169.6 (3CH3CO), 167.0 (C-2), 164.3 (Cpara), 142.9 (C-3), 134.3 (Cipso), 135.2 (C-7a), 131.5 (C-6), 131.3 (2Cmeta), 128.0 (C-8), 126.9 (C-4), 122.9 (C-5), 120.3 (C-3a), 114.1 (2Cortho), 113.8 (C-7), 80.4 (C-1′), 74.0 (C-5′), 70.6 (C-3′), 70.4 (C-2′), 70.1 (C-4′), 55.6 (OCH3), 20.8, 20.8, 20.5 (3CH3CO), 17.6 (C-6′). MS (EI, 70 eV): m/z (%) = 551 (M+, 30), 273 ([M − aglycone]+, 61), 153 (100). HRMS (EI): calc. for C29H29NO10 (M+) 551.17860, found 551.17748.
:ethyl acetate v:v = 3:1) as orange solid (yield: 212 mg, 78%). Mp 160–161 °C (heptane:ethyl acetate).
1H NMR (300 MHz, CDCl3): δ = 8.39 (d (br, overlapped), 1H, H-4), 8.36 (d, 3Jo,m = 8.9 Hz, 2H, 2Hmeta), 8.23 (d, 3Jo,m = 8.9 Hz, 2H, 2Hortho), 7.80 (s, 1H, H-8), 7.53 (d (br), 3J7,6 = 8.1 Hz, 1H, H-7), 7.36 (“dt”, 4J6,4 = 1.2 Hz, 3J6,7 ≈ 3J6,5 ≈ 8.0 Hz, 1H, H-6), 7.03 (“dt”, 4J5,7 = 1.0 Hz, 3J5,4 ≈ 3J5,6 ≈ 7.7 Hz, 1H, H-5), 5.92 (d, 3J1′,2′ = 1.5 Hz, 1H, H-1′), 5.59 (dd, 3J2′,1′ = 1.5 Hz, 3J2′,3′ = 3.0 Hz, 1H, H-2′), 5.30–5.18 (m, 2H, H-3′, H-4′), 3.82–3.71 (m, 1H, H-5′), 2.09, 1.98, 1.84 (3s, 9H, 3CH3CO), 1.36 (d, 3J5′,6′ = 6.2 Hz, 3H, 3H-6′). 13C NMR (63 MHz, CDCl3): δ = 189.1 (C-9), 170.0, 169.7, 169.7 (3CH3CO), 166.7 (C-2), 150.6, 143.9, 142.1, 137.2 (4C), 132.9 (CH), 129.7 (2CH), 127.7, 124.6 (2CH), 124.1 (2CH), 123.2 (CH), 120.0 (C), 114.1 (CH), 80.6, 74.1, 70.5, 70.3, 70.1 (C-1′, C-2′, C-3′, C-4′, C-5′), 20.8, 20.8, 20.5 (3CH3CO), 17.7 (C-6′). MS (EI, 70 eV): m/z (%) = 556 (M+, 12), 294 (9), 273 (63), 153 (80), 111 (72). HRMS (ESI): calc. for C28H27N2O11 ([M + H]+) 567.16094 and for C28H26N2NaO11 ([M + Na]+) 589.14288, found 567.16135 and 589.14317.
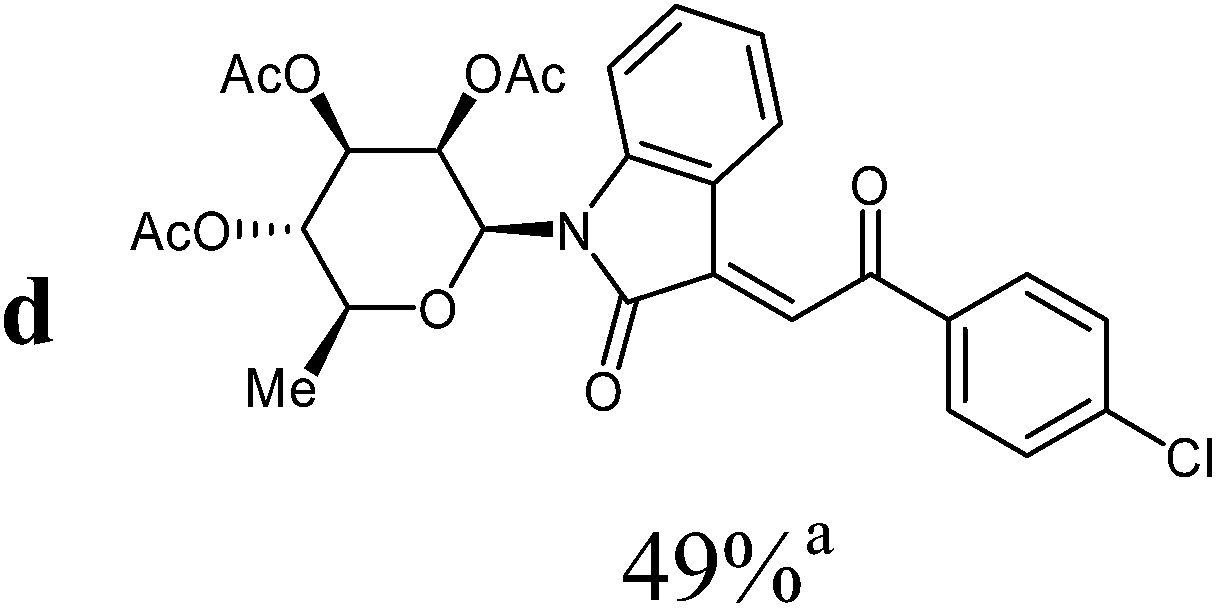
:ethyl acetate v:v = 3:1) as orange solid (yield: 131 mg, 49%). Mp 160–162 °C (heptane:ethyl acetate).
1H NMR (300 MHz, CDCl3): δ = 8.24 (d (br), 3J4,5 = 7.9 Hz, 1H, H-4), 8.02 (“d”, 2H, 3Jo,m = 8.6 Hz, 2Hortho), 7.78 (s, 1H, H-8), 7.51 (d (br, overlapped), 1H, H-7), 7.49 (“d”, 2H, 3Jo,m = 8.6 Hz, 2Hmeta), 7.32 (“dt”, 4J6,4 = 1.3 Hz, 3J6,7 ≈ 3J6,5 ≈ 7.9 Hz, 1H, H-6), 7.00 (“dt”, 4J5,7 = 1.0 Hz, 3J4,5 ≈ 3J5,6 ≈ 7.8 Hz, 1H, H-5), 5.93 (d, 3J1′,2′ = 1.5 Hz, 1H, H-1′), 5.59 (dd, 3J2′,1′ = 1.5 Hz, 3J2′,3′ = 3.0 Hz, 1H, H-2′), 5.29–5.17 (m, 2H, H-3′, H-4′), 3.81–3.71 (m, 1H, H-5′), 2.08, 1.96, 1.82 (3s, 9H, 3CH3CO), 1.34 (d, 3J5′,6′ = 6.2 Hz, 3H, 3H-6′). 13C NMR (75 MHz, CDCl3): δ = 189.6 (C-9), 169.8, 169.6, 169.6 (3CH3CO), 166.7 (C-2), 143.3, 140.3, 135.7, 135.6 (4C), 132.0 (CH), 130.1 (2CH), 129.1 (2CH), 127.1 (CH), 126.1 (CH), 122.9 (CH), 120.0 (C), 113.8 (CH), 80.4, 73.9, 70.5, 70.2, 70.0 (C-1′, C-2′, C-3′, C-4′, C-5′), 20.6, 20.6, 20.4 (3CH3CO), 17.5 (C-6′). MS (EI, 70 eV): m/z (%) = 555 (M+ [35Cl], 22), 557 (M+ [37Cl], 11), 312 (9), 283 (13), 273 (70), 153 (89), 111 (84). HRMS (ESI): calc. for C28H2735ClNO9 ([M + H]+) 556.13689 and for C28H2635ClNaNO9 ([M + Na]+) 578.11883, found 556.13773 and 578.11996.
:ethyl acetate v:v = 3:1) as orange solid (yield: 168 mg, 65%). Mp 200–202 °C (heptane:ethyl acetate).
1H NMR (300 MHz, CDCl3): δ = 8.21 (d (br), 3J4,5 = 7.8 Hz, 1H, H-4), 8.11 (“dd”, 2H, 3Jo,m = 8.9 Hz, 4Jo,F = 5.3 Hz, 2Hortho), 7.79 (s, 1H, H-8), 7.51 (d (br), 3J6,7 = 8.1 Hz, 1H, H-7), 7.31 (“dt”, 4J6,4 = 1.3 Hz, 3J6,7 ≈ 3J6,5 ≈ 7.9 Hz, 1H, H-6), 7.19 (“t”, 2H, 3Jo,m ≈ 3Jm,F ≈ 8.6 Hz, 2Hmeta), 6.99 (“dt”, 4J5,7 = 1.0 Hz, 3J5,4 ≈ 3J5,6 ≈ 7.7 Hz, 1H, H-5), 5.93 (d, 3J1′,2′ = 1.5 Hz, 1H, H-1′), 5.60 (dd, 3J2′,1′ = 1.5 Hz, 3J2′,3′ = 3.0 Hz, 1H, H-2′), 5.29–5.18 (m, 2H, H3′, H-4′), 3.81–3.72 (m, 1H, H-5′), 2.09, 1.98, 1.84 (3s, 9H, 3CH3CO), 1.36 (d, 3J5′,6′ = 6.0 Hz, 3H, 3H-6′). 13C NMR (63 MHz, CDCl3): δ = 189.4 (C-9), 169.9, 169.7, 169.7 (3CH3CO), 166.9 (C-2), 165.5 (d, 1JC,F = 256.3 Hz, Cpara), 143.2, 135.4 (2C), 133.9 (d, 4JC,F = 2.7 Hz, Cipso), 132.0 (CH), 131.6 (d, 3JC,F = 9.6 Hz, 2CHortho), 127.1, 126.6, 123.0 (3CH), 120.1 (C), 116.1 (d, 2JC,F = 22.0 Hz, 2CHmeta), 113.9 (CH), 80.4, 74.0, 70.5, 70.3, 70.1 (C-1′, C-2′, C-3′, C-4′, C-5′), 20.7, 20.7, 20.5 (3CH3CO), 17.6 (C-6′). 19F NMR (282 MHz, CDCl3): δ = 103.34 (s, CF). MS (EI, 70 eV): m/z (%) = 539 (M+, 22), 296 (10), 273 (66), 171 (24), 153 (90), 111 (75). HRMS (EI): calc. for C28H26FNO9 (M+) 539.15861, found 539.15921.
:ethyl acetate v:v = 3:1) as orange solid (yield: 184 mg, 70%). Mp 187–189 °C (heptane:ethyl acetate).
1H NMR (250 MHz, CDCl3): δ = 8.41–8.34 (m, 3H, 2Hmeta, H-4), 8.23 (“d”, 3Jo,m = 8.9 Hz, 2H, 2Hortho), 7.81 (s, 1H, H-8), 7.50 (d (br), 3J6,7 = 8.1 Hz, 1H, H-7), 7.35 (“dt”, 4J4,6 = 1.2 Hz, 3J6,7 ≈ 3J5,6 ≈ 7.9 Hz, 1H, H-6), 7.04 (“dt”, 4J5,7 = 1.0 Hz, 3J4,5 ≈ 3J5,6 ≈ 7.7 Hz, 1H, H-5), 5.97 (d, 3J1′,2′ = 1.5 Hz, 1H, H-1′), 5.61 (dd, 3J2′,1′ = 1.5 Hz, 3J2′,3′ = 3.3 Hz, 1H, H-2′), 5.46–5.28 (m, 2H, H-3′, H-4′), 4.35–4.19 (m, 2H, 2H-6′), 3.93–3.86 (m, 1H, H-5′), 2.11, 2.09, 1.99, 1.84 (4s, 12H, 4CH3CO). 13C NMR (63 MHz, CDCl3): δ = 189.0 (C-9), 170.4, 169.7, 169.6, 169.6 (4CH3CO), 166.7 (C-2), 150.5, 143.6, 142.0, 137.0 (4C), 132.8 (CH), 129.7 (2CH), 127.7, 124.7 (2CH), 124.1 (2CH), 123.2 (CH), 120.0 (C), 114.0 (CH), 80.6, 75.5, 70.5, 69.9, 65.2 (C-1′, C-2′, C-3′, C-4′, C-5′), 62.2 (C-6′), 20.7, 20.7, 20.6, 20.4 (4CH3CO). MS (EI, 70 eV): m/z (%) = 624 (M+, 20), 331 (66), 294 (13), 169 (94), 127 (19), 109 (73). HRMS (EI): calc. for C30H28N2O13 (M+) 624.15859, found 624.16003.
:ethyl acetate v:v = 3:1) as orange solid (yield: 134 mg, 52%). Mp 80–82 °C (heptane:ethyl acetate).
1H NMR (300 MHz, CDCl3): δ = 8.25 (d (br), 3J4,5 = 7.9 Hz, 1H, H-4), 8.02 (“d”, 3Jo,m = 8.6 Hz, 2H, 2Hortho), 7.79 (s, 1H, H-8), 7.47–7.52 (m, 3H, 2Hmeta, H-7), 7.31 (“dt”, 4J4,6 = 1.3 Hz, 3J6,7 ≈ 3J5,6 ≈ 7.9 Hz, 1H, H-6), 7.01 (“dt”, 4J5,7 = 1.0 Hz, 3J4,5 ≈ 3J5,6 ≈ 7.8 Hz, 1H, H-5), 5.97 (d, 3J1′,2′ = 1.5 Hz, 1H, H-1′), 5.61 (dd, 3J2′,1′ = 1.5 Hz, 3J2′,3′ = 3.3 Hz, 1H, H-2′), 5.46–5.29 (m, 2H, H-3′, H-4′), 4.34–4.20 (m, 2H, 2H-6′), 3.92–3.86 (m, 1H, H-5′), 2.11, 2.09, 1.99, 1.84 (4s, 12H, 4CH3CO). 13C NMR (63 MHz, CDCl3): δ = 189.6 (C-9), 170.4, 169.6, 169.5, 169.5 (4CH3CO), 166.8 (C-2), 143.2, 140.4, 135.7, 135.5 (4C), 132.0 (CH), 130.1 (2CH), 129.2 (2CH), 127.2, 126.3, 123.0 (3CH), 120.1 (C), 113.9 (CH), 80.5, 75.4, 70.5, 70.0, 65.3 (C-1′, C-2′, C-3′, C-4′, C-5′), 62.2 (C-6′), 20.7, 20.6, 20.6, 20.4 (4CH3CO). MS (EI, 70 eV): m/z (%) = 613 (M+ [35Cl], 20), 615 (M+ [37Cl], 11), 441 (12), 331 (35), 169 (100), 146 (13), 127 (12), 109 (36). HRMS (ESI): calc. for C30H2935ClNO11 ([M + H]+) 614.14236 and for C30H2835ClNaNO11 ([M + Na]+) 636.12431, found 614.14306 and 636.12571.
:ethyl acetate v:v = 3:1) as orange solid (yield: 158 mg, 63%). Mp 158–160 °C (heptane:ethyl acetate).
1H NMR (300 MHz, CDCl3): δ = 8.21 (d (br), 3J4,5 = 7.8 Hz, 1H, H-4), 8.11 (“dd”, 2H, 3Jo,m = 8.9 Hz, 4Jo,F = 5.4 Hz, 2Hortho), 7.79 (s, 1H, H-8), 7.48 (d (br), 3J6,7 = 8.1 Hz, 1H, H-7), 7.30 (“dt”, 4J4,6 = 1.3 Hz, 3J6,7 ≈ 3J5,6 ≈ 7.9 Hz, 1H, H-6), 7.19 (“t”, 2H, 3Jo,m ≈ 3Jm,F ≈ 8.6 Hz, 2Hmeta), 7.00 (“dt”, 4J5,7 = 1.0 Hz, 3J4,5 ≈ 3J5,6 ≈ 7.7 Hz, 1H, H-5), 5.97 (d, 3J1′,2′ = 1.5 Hz, 1H, H-1′), 5.61 (dd, 3J2′,1′ = 1.5 Hz, 3J2′,3′ = 3.3 Hz, 1H, H-2′), 5.29–5.45 (m, 2H, H-3′, H-4′), 4.34–4.20 (m, 2H, 2H-6′), 3.93–3.86 (m, 1H, H-5′), 2.11, 2.09, 1.98, 1.84 (4s, 12H, 4CH3CO). 13C NMR (75 MHz, CDCl3): δ = 189.4 (C-9), 170.5, 169.7, 169.6, 169.6 (4CH3CO), 166.9 (C-2), 166.3 (d, 1JC,F = 256.8 Hz, Cpara), 143.1, 135.3 (2C), 133.8 (d, 4JC,F = 2.8 Hz, Cipso), 132.0 (CH), 131.6 (d, 3JC,F = 9.5 Hz, 2CHortho), 127.2, 126.8, 123.1 (3CH), 120.1 (C), 116.1 (d, 2JC,F = 22.1 Hz, 2CHmeta), 113.9 (CH), 80.5, 75.5, 70.6, 70.0, 65.3 (C-1′, C-2′, C-3′, C-4′, C-5′), 62.2 (C-6′), 20.8, 20.7, 20.7, 20.5 (4CH3CO). MS (EI, 70 eV): m/z (%) = 597 (M+, 48), 331 (74), 296 (12), 267 (22), 239 (14), 211 (16), 169 (100), 123 (63), 109 (81). HRMS (EI): calc. for C30H28FNO11 [M]+ 597.16409, found 597.16456.
:ethyl acetate v:v = 3:1) as orange solid (yield: 188 mg, 69%). Mp 98–100 °C (heptane:ethyl acetate).
1H NMR (300 MHz, CDCl3): δ = 8.34 (d (br), 3J4,5 = 7.8 Hz, 1H, H-4), 8.18 (“d” (br), 2H, 3Jo,m = 8.2 Hz, 2Hortho), 7.82 (s, 1H, H-8), 7.79 (“d” (br), 2H, 3Jo,m = 8.2 Hz, 2Hmeta), 7.49 (d (br), 3J6,7 = 8.1 Hz, 1H, H-7), 7.33 (“dt”, 4J4,6 = 1.3 Hz, 3J6,7 ≈ 3J5,6 ≈ 7.9 Hz, 1H, H-6), 7.03 (“dt”, 4J5,7 = 1.0 Hz, 3J4,5 ≈ 3J5,6 ≈ 7.8 Hz, 1H, H-5), 5.97 (d, 3J1′,2′ = 1.5 Hz, 1H, H-1′), 5.62 (dd, 3J2′,1′ = 1.5 Hz, 3J2′,3′ = 3.3 Hz, 1H, H-2′), 5.29–5.46 (m, 2H, H-3′, H-4′), 4.20–4.35 (m, 2H, 2H-6′), 3.87–3.93 (m, 1H, H-5′), 2.11, 2.09, 1.99, 1.84 (4s, 12H, 4CH3CO). 13C NMR (75 MHz, CDCl3): δ = 189.7 (C-9), 170.4, 169.6, 169.6, 169.5 (4CH3CO), 166.7 (C-2), 143.4 (C), 140.1 (q, 5JC,F = 1.0 Hz, Cipso), 136.3 (C), 134.8 (q, 2JC,F = 32.7 Hz, Cpara), 132.4 (CH), 129.0 (2CHortho), 127.4 (CH), 125.8 (q, 3JC,F = 3.6 Hz, 2CHmeta), 125.5 (CH), 123.5 (q, 1JC,F = 273.0 Hz, CF3), 123.1 (CH), 120.0 (C), 113.9 (CH), 80.5, 75.4, 70.5, 70.0, 65.2 (C-1′, C-2′, C-3′, C-4′, C-5′), 62.1 (C-6′), 20.7, 20.6, 20.6, 20.4 (4CH3CO). 19F NMR (282 MHz, CDCl3): δ = 113.86 (s, CF3). MS (EI, 70 eV): m/z (%) = 647 (M+, 42), 331 (80), 317 (28), 289 (18), 173 (57), 169 (100), 145 (50), 127 (39), 109 (82). HRMS (EI): calc. for C31H28F3NO11 (M+) 647.16090, found 647.16114.
:ethyl acetate v:v = 3:1) as orange solid (yield: 186 mg, 71%). Mp 205–207 °C (heptane:ethyl acetate).
1H NMR (300 MHz, acetone-d6): δ = 8.37–8.47 (m, 4H, 2Hmeta, 2Hortho), 8.36 (d (br), 3J4,5 = 7.8 Hz, 1H, H-4), 7.88 (s, 1H, H-8), 7.60 (d (br), 3J7,6 = 8.0 Hz, 1H, H-7), 7.51 (“dt”, 4J4,6 = 1.2 Hz, 3J6,7 ≈ 3J5,6 ≈ 7.8 Hz, 1H, H-6), 7.12 (“dt”, 4J5,7 = 1.1 Hz, 3J4,5 ≈ 3J5,6 ≈ 7.7 Hz, 1H, H-5), 5.97 (d, 3J1′,2′ = 9.4 Hz, 1H, H-1′), 5.72 (“t”, 3J2′,1′ ≈ 3J2′,3′ ≈ 9.4 Hz, 1H, H-2′), 5.55 (“t”, 3J4′,3′ ≈ 3J4′,5′ ≈ 9.5 Hz, 1H, H-4′), 5.36 (“t”, 3J3′,2′ ≈ 3J3′,4′ ≈ 9.5 Hz, 1H, H-3′), 4.21–4.35 (m, 3H, H5′, 2H-6′), 2.05, 2.04, 1.96, 1.83 (4s, 12H, 4CH3CO). 13C NMR (63 MHz, acetone-d6): δ = 190.6 (C-9), 170.7, 170.2, 170.1, 169.6 (4CH3CO), 167.7 (C-2), 151.6, 144.0, 143.0, 137.2 (4C), 134.3 (CH), 130.9 (2CH), 128.2, 126.4 (2CH), 124.9 (2CH), 123.9 (CH), 120.8 (C), 113.5 (CH), 80.4, 75.2, 73.9, 68.9, 68.6 (C-1′, C-2′, C-3′, C-4′, C-5′), 62.7 (C-6′), 20.6, 20.6, 20.5, 20.2 (4CH3CO). MS (EI, 70 eV): m/z (%) = 624 (M+, 10), 331 (59), 294 (9), 169 (100), 150 (20), 127 (19), 109 (73). HRMS (EI): calc. for C30H28N2O13 (M+) 624.15859, found 624.15896.
:ethyl acetate v:v = 3:1) as orange solid (yield: 182 mg, 67%). Mp 160–162 °C (heptane:ethyl acetate).
1H NMR (300 MHz, acetone-d6): δ = 8.28–8.32 (m, 3H, 2Hortho, H-4), 7.93 (“d” (br), 2H, 3Jo,m = 8.2 Hz, 2Hmeta), 7.84 (s, 1H, H-8), 7.56 (d (br), 3J7,6 = 8.0 Hz, 1H, H-7), 7.46 (“dt”, 4J4,6 = 1.2 Hz, 3J6,7 ≈ 3J5,6 ≈ 7.8 Hz, 1H, H-6), 7.08 (“dt”, 4J5,7 = 1.1 Hz, 3J4,5 ≈ 3J5,6 ≈ 7.7 Hz, 1H, H-5), 5.95 (d, 3J1′,2′ = 9.4 Hz, 1H, H-1′), 5.70 (“t”, 3J2′,1′ ≈ 3J2′,3′ ≈ 9.4 Hz, 1H, H-2′), 5.53 (“t”, 3J4′,3′ ≈ 3J4′,5′ ≈ 9.5 Hz, 1H, H-4′), 5.33 (“t”, 3J3′,2′ ≈ 3J3′,4′ ≈ 9.5 Hz, 1H, H-3′), 4.18–4.33 (m, 3H, H5′, 2H-6′), 2.02, 2.01, 1.93, 1.80 (4s, 12H, 4CH3CO). 13C NMR (63 MHz, acetone-d6): δ = 190.9 (C-9), 170.7, 170.2, 170.1, 169.6 (4CH3CO), 167.8 (C-2), 143.9 (C), 141.5 (q, 5JC,F = 1.1 Hz, Cipso), 136.9 (C), 134.9 (q, 2JC,F = 32.2 Hz, Cpara), 134.1 (CH), 130.3 (2CHortho), 128.1 (CH), 126.9 (q, 3JC,F = 3.7 Hz, 2CHmeta), 126.7 (CH), 124.9 (q, 1JC,F = 272.5 Hz, CF3), 123.8 (CH), 120.8 (C), 113.5 (CH), 80.4, 75.1, 73.9, 68.9, 68.6 (C-1′, C-2′, C-3′, C-4′, C-5′), 62.7 (C-6′), 20.6, 20.6, 20.5, 20.2 (4CH3CO). 19F NMR (282 MHz, acetone-d6): δ = 113.86 (s, CF3). MS (EI, 70 eV): m/z (%) = 647 (M+, 14), 331 (55), 317 (13), 289 (8), 169 (100), 145 (23), 127 (21), 109 (81). HRMS (EI): calc. for C31H28F3NO11 (M+) 647.16090, found 647.16031.
:ethyl acetate v:v = 3:1) as orange solid (yield: 161 mg, 64%).
1H NMR (300 MHz, acetone-d6): δ = 8.19–8.25 (m, 3H, 2Hortho, H-4), 7.83 (s, 1H, H-8), 7.47–7.58 (m, 2H, H-6, H-7), 7.35 (“t”, 2H, 3Jo,m ≈ 3Jm,F ≈ 8.8 Hz, 2Hmeta), 7.10 (“dt”, 4J5,7 = 1.4 Hz, 3J4,5 ≈ 3J5,6 ≈ 7.6 Hz, 1H, H-5), 5.97 (d, 3J1′,2′ = 9.3 Hz, 1H, H-1′), 5.82 (“t” (br), 3J2′,1′ ≈ 3J2′,3′ ≈ 9.6 Hz, 1H, H-2′), 5.60 (dd, 3J4′,3′ = 3.2 Hz, 3J4′,5′ = 0.8 Hz, 1H, H-4′), 5.46 (dd, 3J3′,2′ = 10.0 Hz, 3J3′,4′ = 3.2 Hz, 1H, H-3′), 4.58 (ddd, 3J5′,4′ = 0.8 Hz, 3J5′,6a′ = 5.6 Hz, 3J5′,6b′ = 6.8 Hz, 1H, H-5′), 4.29 (dd, 3J6a′,5′ = 5.6 Hz, 2J6a′,6b′ = 11.5 Hz, 1H, H-6a′), 4.14 (dd, 3J6b′,5′ = 6.8 Hz, 2J6b′,6a′ = 11.5 Hz, 1H, H-6b′), 2.31, 1.98, 1.95, 1.87 (4s, 12H, 4CH3CO). 13C NMR (75 MHz, acetone-d6): δ = 190.3 (C-9), 170.6, 170.5, 170.2, 169.8 (4CH3CO), 167.7 (C-2), 167.0 (d, 1JC,F = 254.2 Hz, Cpara), 143.7, 136.1 (2C), 135.1 (d, 4JC,F = 2.8 Hz, Cipso), 133.6, (CH), 132.6 (d, 3JC,F = 9.9 Hz, 2CHortho), 127.9, 127.6, 123.7 (3CH), 120.9 (C), 116.9 (d, 2JC,F = 22.2 Hz, 2CHmeta), 113.1 (CH), 80.5, 73.8, 72.0, 68.5, 66.4 (C-1′, C-2′, C-3′, C-4′, C-5′), 62.6 (C-6′), 20.7, 20.6, 20.5, 20.3 (4CH3CO). 19F NMR (282 MHz, acetone-d6): δ = 71.74 (s, CF). MS (EI, 70 eV): m/z (%) = 597 (M+, 30), 331 (94), 317 (13), 289 (12), 211 (11), 169 (98), 146 (17), 127 (26), 123 (60), 109 (68). HRMS (ESI): calc. for C30H29FNO11 ([M + H]+) 598.17192, found 598.17140.
:ethyl acetate v:v = 3:1) as orange solid (yield: 180 mg, 66%). Mp 91–93 °C (heptane:ethyl acetate).
1H NMR (300 MHz, acetone-d6): δ = 8.33–8.37 (m, 3H, 2Hortho, H-4), 7.97 (d (br), 3Jo,m = 8.2 Hz, 2H, 2Hmeta), 7.88 (s, 1H, H-8), 7.51–7.60 (m, 2H, H-6, H-7), 7.11–7.17 (m, 1H, H-5), 5.97 (d, 3J1′,2′ = 9.2 Hz, 1H, H-1′), 5.82 (“t” (br), 3J2′,1′ ≈ 3J2′,3′ ≈ 9.6 Hz, 1H, H-2′), 5.60 (dd, 3J4′,3′ = 3.3 Hz, 3J4′,5′ = 0.9 Hz, 1H, H-4′), 5.46 (dd, 3J3′,2′ = 10.0 Hz, 3J3′,4′ = 3.3 Hz, 1H, H-3′), 4.59 (ddd, 3J5′,4′ = 0.9 Hz, 3J5′,6a′ = 5.7 Hz, 3J5′,6b′ = 6.8 Hz, 1H, H-5′), 4.29 (dd, 3J6a′,5′ = 5.7 Hz, 2J6a′,6b′ = 11.5 Hz, 1H, H-6a′), 4.15 (dd, 3J6b′,5′ = 6.8 Hz, 2J6b′,6a′ = 11.5 Hz, 1H, H-6b′), 2.32, 1.99, 1.96, 1.87 (4s, 12H, 4CH3CO). 13C NMR (63 MHz, acetone-d6): δ = 190.9 (C-9), 170.6, 170.5, 170.2, 169.8 (4CH3CO), 167.7 (C-2), 144.0, 141.5, 137.0 (3C), 134.9 (q, 2JC,F = 32.3 Hz, Cpara), 134.1 (CH), 130.3 (2CHortho), 128.2 (CH), 126.9 (q, 3JC,F = 3.8 Hz, 2CHmeta), 126.7 (CH), 124.8 (q, 1JC,F = 272.0 Hz, CF3), 123.8 (CH), 120.9 (C), 113.2 (CH), 80.5, 73.8, 72.0, 68.5, 66.4 (C-1′, C-2′, C-3′, C-4′, C-5′), 62.6 (C-6′), 20.7, 20.6, 20.5, 20.3 (4CH3CO). 19F NMR (282 MHz, CDCl3): δ = 113.85 (s, CF3). MS (EI, 70 eV): m/z (%) = 647 (M+, 18), 331 (96), 317 (16), 289 (13), 169 (96), 145 (20), 127 (20), 109 (56). HRMS (EI): calc. for C31H28F3NO11 (M+) 647.16090, found 647.16008.
Material and methods for the bioactivity tests
Acknowledgements
Financial support by the State of Mecklenburg-Western Pommerania, the Deutsche Forschungsgemeinschaft and the Deutsche Krebshilfe (Melanomverbund, grant numbers 108008 TP5 and TP7) are gratefully acknowledged. Analysis of data from the kinase ATP binding assays was supported by F. Gaunitz, Clinic for Neurosurgery, University of Leipzig.References
- Z. Xiao, Y. Hao, B. Liu and L. Qian, Leuk. Lymphoma, 2002, 43, 1763–1768 CAS.
- (a) R. Hoessel, S. Leclerc, J. A. Endicott, M. E. M. Nobel, A. Lawrie, P. Tunnah, M. Leost, E. Damiens, D. Marie, D. Marko, E. Niederberger, W. Tang, G. Eisenbrand and L. Meijer, Nat. Cell Biol., 1999, 1, 60–67 CrossRef CAS PubMed; (b) M. J. Moon, S. K. Lee, J.-W. Lee, W. K. Song, S. W. Kim, J. I. Kim, C. Cho, S. J. Choi and Y.-C. Kim, Bioorg. Med. Chem., 2006, 14, 237–246 CrossRef CAS PubMed.
- (a) E. A. Sausville, D. Zaharevitz, R. Gussio, L. Meijer, M. Louarn-Leost, C. Kunick, R. Schultz, T. Lahusen, D. Headlee, S. Stinson, S. G. Arbuck and A. Senderowicz, Pharmacol. Ther., 1999, 82, 285–292 CrossRef CAS; (b) P. M. Fischer and D. P. Lane, Curr. Med. Chem., 2000, 7, 1213–1245 CrossRef CAS.
- A. Beauchard, Y. Ferandin, S. Frere, O. Lozach, M. Blairvacq, L. Meijer, V. Thiery and T. Besson, Bioorg. Med. Chem., 2006, 14, 6434–6443 CrossRef CAS PubMed.
- Review: G. Karapetyan, K. Chakrabarty, M. Hein and P. Langer, ChemMedChem, 2011, 6, 25–37 CrossRef CAS PubMed.
- (a) M. Sassatelli, E. Saab, F. Anizon, M. Prudhomme and P. Moreau, Tetrahedron Lett., 2004, 45, 4827–4830 CrossRef CAS PubMed; (b) M. Sassatelli, F. Bouchikhi, S. Messaoudi, F. Anizon, E. Debiton, C. Barthomeuf, M. Prudhomme and P. Moreau, Eur. J. Med. Chem., 2006, 41, 88–100 CrossRef CAS PubMed; (c) M. Sassatelli, F. Bouchikhi, B. Aboab, F. Anizon, F. Doriano, M. Prudhomme and P. Moreau, Anti-Cancer Drugs, 2007, 18, 1069–1074 CrossRef CAS PubMed; (d) L. Wang, X. Liu and R. Chen, US Pat. 6,566,341, 2003, Chem. Abstr., 2003, 138, 379213; (e) L. Wang, X. Liu and R. Chen, WO Patent 03051900, 2003, Chem. Abstr., 2003, 139, 47135.
- (a) S. Libnow, M. Hein, D. Michalik and P. Langer, Tetrahedron Lett., 2006, 47, 6907–6909 CrossRef CAS PubMed; (b) S. Libnow, K. Methling, M. Hein, D. Michalik, M. Harms, K. Wende, A. Flemming, M. Koeckerling, H. Reinke, P. J. Bednarski, M. Lalk and P. Langer, Bioorg. Med. Chem., 2008, 16, 5570–5583 CrossRef CAS PubMed.
- (a) F. Erben, D. Kleeblatt, M. Sonneck, M. Hein, H. Feist, T. Fahrenwaldt, C. Fischer, A. Matin, J. Iqbal, M. Ploetz, J. Eberle and P. Langer, Org. Biomol. Chem., 2013, 11, 3963–3978 RSC; (b) D. Kleeblatt, B. Siyo, M. Hein, V. O. Iaroshenko, J. Iqbal, A. Villinger and P. Langer, Org. Biomol. Chem., 2013, 11, 886–895 RSC; (c) M. Kunz, K. M. Driller, M. Hein, S. Libnow, I. Hohensee, R. Ramer, B. Hinz, A. Berger, J. Eberle and P. Langer, ChemMedChem, 2010, 5, 534–539 CrossRef CAS PubMed; (d) S. Libnow, M. Hein and P. Langer, Synlett, 2009, 221–224 CAS; (e) S. Libnow, M. Hein and P. Langer, Tetrahedron Lett., 2008, 49, 289–291 CrossRef CAS PubMed; (f) D. Kleeblatt, C. A. Cordes, P. Lebrenz, M. Hein, H. Feist, A. Matin, R. Raza, J. Iqbal, O. Munshi, Q. Rahman, A. Villinger and P. Langer, RSC Adv., 2014, 4, 22828 RSC; (g) F. Erben, D. Michalik, H. Feist, D. Kleeblatt, M. Hein, A. Matin, J. Iqbal and P. Langer, RSC Adv., 2014, 4, 10879 RSC.
- (a) C. L. Woodard, Z. Li, A. K. Kathcart, J. Terrell, L. Gerena, M. Lopez-Sanchez, D. E. Kyle, A. K. Bhattacharjee, D. A. Nichols, W. Ellis, S. T. Prigge and J. A. Geyer, J. Med. Chem., 2003, 46, 3877–3882 CrossRef CAS PubMed; (b) R. S. Belenkaya, E. I. Boreko, M. N. Zemtsova, M. I. Kalinina and M. M. Timofeeva, Pharm. Chem. J., 1981, 15, 171–176 CrossRef; (c) J. Strigacova, D. Hudecova, M. Mikulasova, L. Varecka, A. Lasikova and D. Vegh, Folia Microbiol., 2001, 46, 187–192 CrossRef CAS; (d) J. W. Lockman, M. D. Reeder, R. Robinson, P. A. Ormonde, D. M. Cimbora, B. L. Williams and J. A. Willardsen, ACS Chem. Biol., 2009, 4, 199–208 CrossRef PubMed; (e) E. N. Koz'minykh, E. S. Berezina, V. E. Kolla, S. A. Shelenkova, E. V. Voronina and V. O. Koz'minykh, Khim.–Farm. Zh., 1997, 31, 31–36 Search PubMed.
- (a) R. Stollé, Chem. Ber., 1913, 46, 3915–3916 CrossRef; (b) J. F. M. da Silva, S. J. Garden and A. C. Pinto, J. Braz. Chem. Soc., 2001, 12, 273–324 CrossRef CAS PubMed; (c) M. N. Preobrazhenskaya, I. V. Yartseva and L. V. Ektova, Dokl. Chem., 1974, 215, 219–222 Search PubMed; (d) C. Chavis, C. De Gourcy, F. Dumont and J.-L. Imbach, Carbohydr. Res., 1983, 113, 1–20 CrossRef CAS; (e) J. G. Douglas and J. Honeyman, J. Chem. Soc., 1955, 3674–3680 RSC; (f) M. N. Preobrazhenskaya, I. V. Yartseva and L. V. Ektova, Nucleic Acid Chem., 1978, 2, 725–727 Search PubMed; (g) I. V. Yartseva, L. V. Ektova and M. N. Preobrazhenskaya, Bioorg. Khim., 1975, 1, 189–194 CAS.
- ESI.†.
- G. Zemplén, A. Gerecs and I. Hadácsy, Chemische Berichte, 1936, 69, 1827–1829 CrossRef.
- J. Brüggen and C. Sorg, Cancer Immunol. Immunother., 1983, 15, 200–205 CrossRef.
- A. Berger, S. A. Quast, M. Plötz, M. Hein, M. Kunz, P. Langer and J. Eberle, Biochem. Pharmacol., 2011, 81, 71–81 CrossRef CAS PubMed.
- S. A. Quast, A. Berger, M. Plötz and J. Eberle, Eur. J. Cell Biol., 2014, 93, 42–48 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1019590. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra14301a |
| This journal is © The Royal Society of Chemistry 2015 |