
Adsorption behavior of Cr(vi) from aqueous solution onto magnetic graphene oxide functionalized with 1,2-diaminocyclohexanetetraacetic acid
Abstract
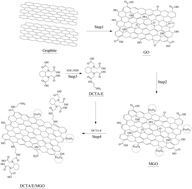
A novel magnetic composite adsorbent was synthesized by grafting 1,2-diaminocyclohexanetetraacetic acid to magnetic graphene oxide (DCTA/E/MGO). The DCTA/E/MGO was employed for removing Cr(VI) from aqueous solution in this study. The composite was characterized by FESEM, TEM, BET, XRD, FT-IR and XPS. The adsorption behaviors of Cr(VI) by DCTA/E/MGO in aqueous solution were systematically investigated. Second order kinetic and Freundlich isotherm models validated the experimental data. The adsorption rate was influenced by both film diffusion and intraparticle diffusion. Thermodynamic parameters revealed that the adsorption reaction was an endothermic and spontaneous process. The novel adsorbent exhibited better Cr(VI) removal efficiency in solutions with low pH. The decontamination of Cr(VI) by DCTA/E/MGO was influenced by ionic strength. These results are important for estimating and optimizing the removal of metal ions by the DCTA/E/MGO composite.
 Please wait while we load your content...
Please wait while we load your content...

