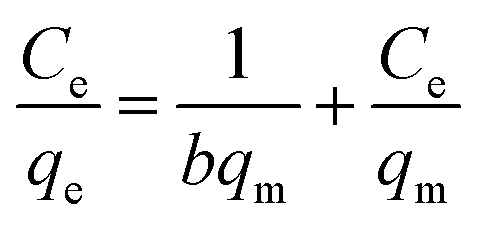DOI:
10.1039/C5RA01815C
(Paper)
RSC Adv., 2015,
5, 32272-32282
Poly(acrylic acid) functionalized magnetic graphene oxide nanocomposite for removal of methylene blue
Received
29th January 2015
, Accepted 30th March 2015
First published on 30th March 2015
Abstract
A polyacrylic acid (PAA) functionalized magnetic Fe3O4 nanoparticle-graphene oxide nanocomposite (PAA/MGO) was synthesized by a facile method. The structure and surface properties of MGO and PAA/MGO composites were characterized by infrared (IR) spectroscopy, X-ray photoelectron spectroscopy (XPS), high resolution transmission electron microscopy (HRTEM), thermogravimetric analysis (TGA) and zeta potential measurements. The adsorption of a model dye pollutant, methylene blue (MB), on MGO and PAA/MGO was investigated in batch tests. The functionalization of PAA to MGO significantly enhances the maximum adsorption capacity of MB (at pH = 7) from ∼70 mg g−1 (on MGO) to ∼291 mg g−1 (PAA/MGO). The adsorption of MB on MGO and PAA/MGO was mainly driven by the electrostatic attraction between positively charged MB molecules and negatively charged nanocomposite surfaces, and the higher adsorption capacity of PAA/MGO is mainly attributed to the functionalization of PAA and its higher content of charged carboxyl groups than MGO. The adsorption capacity of MB on both MGO and PAA/MGO adsorbents also increases with increasing solution pH from 3 to 11, due to enhanced electrostatic attraction at high pH conditions. The limited adsorption capacity of MB on MGO and PAA/MGO at pH 3, when electrostatic attraction is almost negligible, indicates that π–π interaction between the GO surface and MB also plays a role in the adsorption process. The PAA/MGO shows a rapid adsorption rate and high adsorption capacity of MB with magnetic properties for easy separation and excellent recyclability, which endows the nanocomposite with great potential for the removal of cationic organic pollutants in wastewater treatment.
1. Introduction
Dyes are widely used in many industries like textile, paper and pulp, dyestuffs and plastics industries. Dye contaminants extensively exist in wastewater discharged by miscellaneous industrial activities which cause various environmental problems.1,2 A trace amount of water-soluble dyes can cause noticeable colorization of water, which will reduce the light penetration and thus interfere with the photosynthetic activity of aquatic organisms.3,4 Moreover, dyes are generally toxic to living creatures and cannot be easily degraded by microorganisms.1,2,5 Therefore, economic techniques to efficiently remove dye contaminants from wastewater are highly desirable. To date, various technologies have been invented and applied for the removal of dyes from wastewater, including coagulation/flocculation,6,7 photocatalysis,8–10 ultrafiltration,11,12 and adsorption.13,14 Among these methods, adsorption has received much attention due to its high efficiency, simplicity and economy.13,15–17 A variety of adsorbents such as clay,18 zeolite19 and carbon materials,20 have been developed to remove colored organic pollutants. Yet, challenging issues like relatively low removal efficiency and separation difficulty still remain for most conventional adsorbents.
Graphene oxide (GO) has unique 2-D structure with high surface area and rich functional groups, such as carboxylic, epoxide and hydroxide groups, which endow its good dispersibility in water and availability for further modification,21,22 attracting considerable attention for wastewater treatment.23–29 Inherently, the basal plane of GO is able to interact with the aromatic rings of organics by π–π stacking,30,31 contributing to its adsorption of organic dye pollutants. To expand the practical applicability of GO, various chemical and physical modifications have been applied to decorate GO with specific functionalities, such as photocatalysis ability,10 improved adsorption capacity for specific pollutants32 and recyclability.14 Magnetic particles have also been introduced to GO sheets to enhance the separation efficiency.33–36 It is noted, however, the introduction of magnetic particles to GO commonly leads to a relatively low adsorption capacity of the composite materials. One possible way to cope with this dilemma is to graft the magnetic graphene oxide (MGO) with functional polymers, which contain abundant functional groups such as carboxyl and amino groups and can adsorb pollutants through electrostatic interaction or chelation.37 Although much effort has been devoted to the fabrication and characterization of graphene–polymer composites, most of the attempts focused on their mechanical and electric performance instead of the adsorptive property in wastewater treatment.38,39 The role of polymers in most MGO–polymer composites reported was usually to strengthen the mechanical property of the composites.40,41 Among the limited studies on polymer modified graphene oxide targeting dye removal, cellulose and chitosan have been chosen as the functional polymers to eliminate methylene blue (MB) and fuchsine from wastewater, respectively.42,43 Functionalization of MGO composites using polymers containing abundant carboxylic groups (like polyacrylic acid), which have strong affinity to positively charged organic pollutants and the ability to enhance water dispersibility, has not been reported.
Herein, we report a facile and effective method to prepare polyacrylic acid (PAA) modified magnetic graphene oxide composite (PAA/MGO). Magnetic graphene oxide composite (MGO) was first synthesized using a co-precipitation method,21 and PAA was then introduced to the as-prepared MGO through carbodiimide activation under sonication at room temperature.44–47 The carboxylic groups of PAA can be bound to GO surfaces, and also have electrostatic affinity to cationic pollutants, which is expected to synergistically enhance the adsorption performance.48 In this work, PAA/MGO was utilized to remove organic dye pollutants (i.e. methylene blue), and a higher adsorption capacity with excellent recyclability was achieved as compared to that of magnetic graphene oxide or graphene oxide composites reported previously.33–35,49–51 The adsorption mechanism was also investigated and proposed.
2. Materials and experiment methods
2.1. Chemicals and materials
FeCl3·6H2O and FeCl2·4H2O were purchased from Sigma-Aldrich. KMnO4, NaNO3, methylene blue (MB), NaOH, HCl, ammonium hydroxide and hydrogen peroxide were purchased from Fisher Scientific. Graphite flake (325 mesh), H2SO4 (98%), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) and polyacrylic acid (25%, average MW 240![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were obtained from Alfa Aesar. All chemicals used in this study were analytical grade and used as received.
000) were obtained from Alfa Aesar. All chemicals used in this study were analytical grade and used as received.
2.2. Preparation of graphene oxide (GO)
A modified Hummer's method was applied to synthesize GO.52 Briefly, graphite (2 g) and NaNO3 (1.5 g) were mixed in a 250 mL three-necked flask in an ice bath, and H2SO4 (98%, 150 mL) was added into the mixture with stirring. Then, KMnO4 (9 g) was slowly added to the mixture over about 1 h. Stirring and ice bath were maintained during the addition of KMnO4, which were kept for another 2 h after the addition. The ice bath was removed afterwards while the stirring was kept vigorously for 5 days at room temperature. H2O2 solution (6 mL) was then added into the mixture with another 2 hours agitation. Sequentially, 250 mL solution of deionization (DI) water mixed with H2SO4 (98%, 7.5 mL) and H2O2 (30 wt%, 4.17 mL) was added to dilute and wash the mixture. The resulted mixture was thoroughly washed by DI water, centrifuged, and then dialyzed for 5 days. Finally, the GO aqueous solution was lyophilized and fluffy black dried GO was obtained.
2.3. Preparation of magnetic graphene oxide (MGO)
MGO was synthesized following a modified co-precipitation method.53 The procedure of a typical synthesis experiment is as follows. Firstly, 100 mg GO was dispersed into 100 mL DI water by ultrasonication until fully dispersed. Then the GO solution was added into a 250 mL three-necked flask and stirred vigorously. Secondly, FeCl3·6H2O (0.819 g) and FeCl2·4H2O (0.294 g) were dissolved in DI water (25 mL), and the solution was dropped into the GO solution in the flask under severe agitation for 20 min. Afterwards, the mixture was heated to 50 °C, and the pH was adjusted to 10 with the observation of black precipitate. The solution was further heated to 85 °C and the heat was maintained for an hour. The as-prepared MGO was magnetically separated and washed for more than 3 times. Finally, black MGO powder was obtained by freeze-drying.
2.4. Preparation of PAA/MGO
PAA/MGO composite was synthesized based on a modified synthetic method for PAA/Fe3O4.37 Briefly, MGO (50 mg) was dispersed in 1 mL of buffer (3 mM phosphate, pH = 6.0, 0.1 M NaCl) by sonication in a centrifuge tube. Afterwards, 0.25 mL of carbodiimide solution (0.025 g mL−1 in buffer) was added to the MGO solution. After 10 min sonication, 0.286 mL of PAA solution (25 wt%.) was mixed with 0.963 mL buffer. The PAA mixture was then added to the MGO mixture. The reaction was carried out under sonication for another 30 min. Finally, PAA/MGO was magnetically separated and washed by DI water for over 3 times. Grey powder of PAA/MGO was obtained after freeze-drying.
2.5. Sample characterizations
Fourier transform infrared (FTIR) spectra were recorded on a Thermo Nicolet 8700 FTIR Spectrometer. Thermogravimetric analysis (TGA) was carried out using a TA instruments Q500 under Ar flow (5.0 mL min−1) and air (200.0 mL min−1) at a heating rate of 10 °C min−1. Transmission electron microscopy (TEM) images were recorded by CM20 FEG transmission electron microscope operated at 200 kV. Atomic force microscopy (AFM) imaging was conducted using an Asylum MFP-3D AFM to characterize the morphology and thickness of GO in tapping mode. GO sample was prepared by dropping GO aqueous solution on a freshly cleaved mica surface, followed by overnight drying. X-Ray photoelectron spectroscopy (XPS) measurements were carried out on a Kratos Axis Ultra spectrometer using Al Ka radiation, operated at 12 mA, 14 kV, of which the peak fitting was performed by CasaXPS software. Zeta potential was measured using a Malvern Nanosizer Nano ZSP. The concentrations of MB in aqueous solutions during adsorption tests were characterized using an Evolution 300 UV-Vis spectrophotometer (Thermo Fisher Scientific). The magnetic hysteresis measurements were conducted at room temperature under a maximum applied field of 60000 Oe on a Quantum Design 9T-PPMS magnetometer.
2.6. Adsorption tests
The adsorption of MB in aqueous solutions using MGO and PAA/MGO composites was carried out in batch experiments. Solutions of adsorbents (4 mg) dispersed in deionized (DI) water (0.5 mL) were mixed with different concentrations of MB solutions (25 mL) in centrifuge tubes (pH = 7, pH was adjusted using 0.1 M HCl and 0.1 NaOH). The adsorption experiments were performed by putting the centrifuge tubes in a shaker (300 rpm) and monitored with time. Portions of the mixture were taken out at different time intervals, of which the adsorbents were magnetically removed and the residue concentrations of MB were determined with UV-visible spectrophotometer.33,54 The adsorption kinetics and isotherms of MB on the as-prepared adsorbents were obtained under aforementioned condition.
The effect of pH on the adsorption of MB on MGO and PAA/MGO was also investigated. Solutions of MGO or PAA/MGO (4 mg) dispersed in DI water (0.5 mL) and 20 mg L−1 MB solutions (25 mL) were mixed and adjusted to different pH (pH = 3, 5, 7, 9, 11). The mixtures were left in shaker (300 rpm) for 24 h to reach adsorption equilibrium. The adsorption capacity qt (mg g−1) and removal percentage p% can be calculated by eqn (1)33 and (2),55 where C0 and Ct (mg L−1) are the initial concentration and concentration at time t of residual MB in the solutions, respectively, m (g) stands for the mass of the adsorbent used, V0 (mL) and Vt (mL) are initial volume and volume at time t of solutions, respectively.
| |
 | (1) |
| |
 | (2) |
2.7. Zeta potential measurements
MGO or PAA/MGO (5 mg) was added into 10 mM NaCl solutions (10 mL) and the suspensions were sonicated until fully dispersed. The pH of the supernatants of the suspensions was adjusted to pH ∼ 3 to 11 using 0.1 M NaOH or HCl, and the zeta potential was then determined using a Malvern Nanosizer Nano ZSP. The measurements for each type of samples were repeated for 3 times.
2.8. Recyclable usage
MGO and PAA/MGO (8 mg) aqueous solutions (0.5 mL) were added to 20 mg L−1 MB solutions (25 mL) respectively and then the pH of the mixtures was adjusted to pH ∼ 7 using 0.1 M NaOH or HCl. The solutions were then left in the shaker (300 rpm) for 5 h. After adsorption, the adsorbents were collected by external magnet and MB concentrations in the residue solutions were measured by UV spectroscopy. For desorption, the recycled adsorbents were dispersed in 10 mL ethanol solution (5% vol acetic acid), sonicated for 15 min and recovered magnetically. The desorption process was repeated for 3 times. After magnetic separation, the adsorbents were washed with 2 mL DI water and then magnetically separated for reuse.
3. Results and discussion
3.1. Synthesis and characterizations of nanocomposite adsorbents
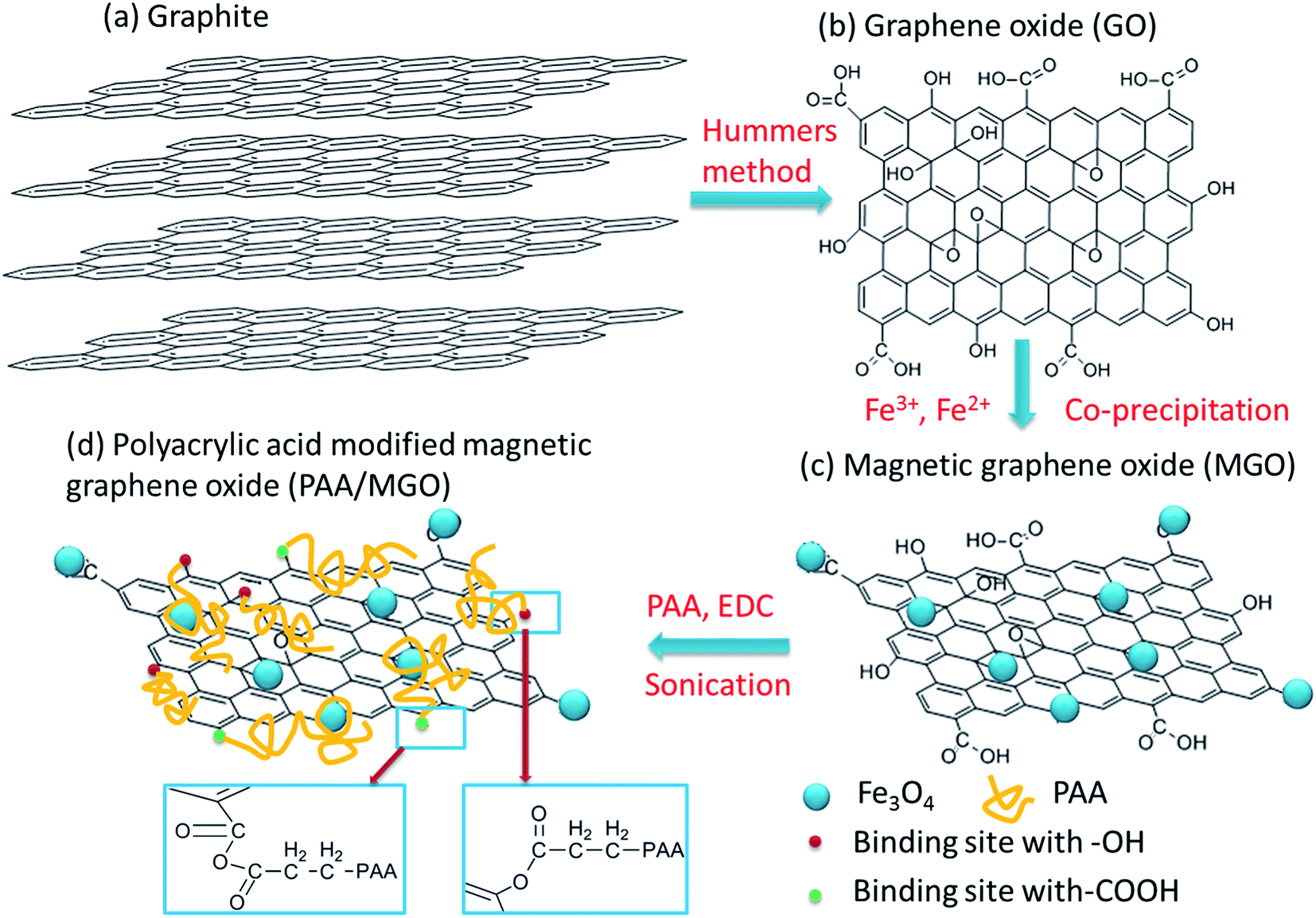
Fig. 1 shows the synthesis route of PAA/MGO composite. GO was first prepared from graphite based on a modified Hummers method.52 FeCl2 and FeCl3 solution (Fe2+:Fe3+ = 2:1) was dropped to GO solutions as iron source, with Fe2+ and Fe3+ ions attracted to the negative-charged oxygen atoms of GO. The solution was adjusted to alkaline condition (pH = 10) and Fe3O4 particles were formed in situ by co-precipitation.56 PAA was then bound to the surfaces of GO by a “grafting from” method.37,57
 |
| | Fig. 1 Schematic of synthesis process for PAA/MGO composite. Note, the binding of PAA and –COOH groups on GO by forming acid anhydride groups as illustrated in (d) may not be very stable in aqueous solution, and the binding between PAA and –OH groups on GO would dominate the grafting interaction. | |
AFM topography and height profile of GO are shown in Fig. 2a and b, respectively, indicating a single layer of GO sheet with thickness of ∼0.91 nm which is consisted with literature value.58 Single layer of GO sheet is thicker than pristine graphene monolayer that is atomically flat with a well-known van der Waals thickness of ∼0.34 nm, which is mainly due to the covalently bonded groups on the carbon atoms and distorted sp3-hybridized geometry transformed from the planar sp2-hybridized geometry.55 The single layer structure of GO obtained contributes to its high surface area allowing further surface modification.
 |
| | Fig. 2 (a) AFM topographic image of GO sheets, and (b) height profile for monolayer GO. | |
The TEM image of GO (Fig. 3a) reveals that GO possesses a transparent sheet structure. TEM image of MGO (Fig. 3b) shows that the magnetic nanoparticles (with an average diameter of ∼7 nm) disperse evenly on the transparent graphene oxide sheets and have a crystal structure with a lattice spacing of 0.48 nm, corresponding to the (111) plane of spinel-structured iron oxide, as shown in the HRTEM image of MGO (Fig. 3d).59 The modification of PAA on MGO shows no significant change of the morphology of iron oxide nanoparticles on graphene oxide sheets in TEM image of PAA/MGO (Fig. 3c). It is noted that after the binding of PAA on MGO, the crystal structure of the magnetic nanoparticles on PAA/MGO is much less visible in the HRTEM image (Fig. 3e) as compared to that in the HRTEM image of MGO (Fig. 3d), which is most likely due to the electron scattering and energy loss in the grafted PAA.60–62 Powder X-ray diffraction (XRD) was employed to characterize the crystalline structure of magnetic iron oxide nanoparticles in the MGO and PAA/MGO composites, and the XRD patterns are shown in Fig. 4a and are in accordance with the standard XRD spectra for Fe3O4 (PDF#01-071-6336). The XRD results confirm the successful deposition of Fe3O4 nanoparticles on GO sheets and that the addition of PAA does not change the crystalline structure of these nanoparticles.
 |
| | Fig. 3 TEM images of (a) GO, (b) MGO and (c) PAA/MGO, HRTEM images of (d) MGO, and (e) PAA/MGO. | |
 |
| | Fig. 4 (a) XRD patterns of MGO and PAA/MGO, (b) FTIR spectra of GO, PAA, MGO and PAA/MGO. | |
The FTIR spectra of GO, PAA, MGO and PAA/MGO are shown in Fig. 4b. For the FTIR spectrum of GO, the peaks from 1000 to 1300 cm−1 can be ascribed to the C–O vibration of hydroxyl groups and C–O–C vibration of epoxy groups; the peaks around 3436 and 3625 cm−1 can be attributed to the O–H vibration of hydroxyl groups and the peak at 1739 cm−1 is due to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration of carboxylic groups,63,64 indicating that the oxidized product of graphite (i.e. GO) via the modified Hummers' method possesses various oxygen-containing functional groups. With the deposition of Fe3O4 nanoparticles on GO sheets, the intensity of the peaks for the hydroxyl, epoxy and carboxylic groups was weakened, which is most likely because the oxygen of these groups can interact with iron ions to form oxide. For PAA/MGO, the appearance of –CH2 vibration at 1455 and 2923 cm−1 and the enhanced –CO vibration peak of carboxylic groups confirm the deposition of PAA on the composite surfaces.65
O vibration of carboxylic groups,63,64 indicating that the oxidized product of graphite (i.e. GO) via the modified Hummers' method possesses various oxygen-containing functional groups. With the deposition of Fe3O4 nanoparticles on GO sheets, the intensity of the peaks for the hydroxyl, epoxy and carboxylic groups was weakened, which is most likely because the oxygen of these groups can interact with iron ions to form oxide. For PAA/MGO, the appearance of –CH2 vibration at 1455 and 2923 cm−1 and the enhanced –CO vibration peak of carboxylic groups confirm the deposition of PAA on the composite surfaces.65
The C1s XPS spectra of GO, MGO and PAA/MGO are shown in Fig. 5a–c, respectively. The spectra were calibrated using the binding energy of the adventitious C1s at 284.6 eV. In Fig. 5, the deconvolution of C1s XPS spectra of GO, MGO and PAA/MGO shows four peaks at 284.6 eV, 286.5 eV, 287.7 eV and 288.3 eV, which can be assigned to saturated C–C bonding of graphene, the carbon-linking hydroxyl groups (C–OH) and epoxy groups (C–O–C), carbonyl groups (CO), and carboxyl groups (–COOH), respectively.66,67 The bond percentage and binding energy (B. E.) of different bonds based on the C1s spectra for GO, MGO and PAA/MGO are summarized in Table 1. Table 1 shows that after the co-precipitation of magnetic particles, the relative intensity of peaks corresponding to oxygen-containing groups significantly decreases, agreeing with FTIR results, which supports the hypothesis that the oxygen-containing groups reacted with iron ions to form Fe3O4 nanoparticles. Compared to MGO, the intensity of CO peak of PAA/MGO dramatically increases, suggesting the successful deposition of PAA on MGO.
 |
| | Fig. 5 C1s XPS spectra of (a) GO, (b) MGO and (c) PAA/MGO. | |
Table 1 XPS C1s peak information for four types of C bonds in GO, MGO and PAA/MGO
| Materials |
COOH |
CO |
C–O |
C–C |
| % |
B.E. |
% |
B.E. |
% |
B.E. |
% |
B.E. |
| GO |
7.4 |
288.4 |
4.29 |
287.3 |
44.5 |
286.6 |
43.9 |
284.6 |
| MGO |
5.6 |
288.7 |
2.11 |
287.7 |
31.2 |
286.4 |
61.1 |
284.6 |
| PAA/MGO |
13.8 |
288.7 |
2.11 |
287.9 |
25.8 |
286.4 |
58.3 |
284.6 |
The thermal behaviors of GO, MGO, PAA/MGO and Fe3O4 were characterized by thermogravimetric analysis and the TGA results are shown in Fig. 6. For the TGA curve of GO, the weight loss below 120 °C is due to the loss of physically adsorbed water. The weight loss from 120 °C to ∼300 °C is ascribed to the loss of oxygen-containing functional groups. The final weight loss from 430 °C to 530 °C is mainly attributed to the burning of carbon.68 For Fe3O4, a slight weight gain below 200 °C is due to the oxidization of Fe3O4 to γ-Fe2O3.34 In contrast, the weight gain of MGO and PAA/MGO owing to the oxidization of Fe3O4 was overwhelmed by the decomposition of oxygen-containing groups, thus these two composites showed gradual weight loss with increasing temperature till 400 °C. Based on the mass loss of the composite materials in TGA tests shown in Fig. 6, the mass fraction of Fe3O4 in MGO was evaluated to be ∼71% and the mass fraction of PAA in PAA/MGO composite was estimated as ∼15%.
 |
| | Fig. 6 TGA curves of GO (black), MGO (blue), PAA/MGO (green) and Fe3O4 (red). | |
3.2. Magnetization tests
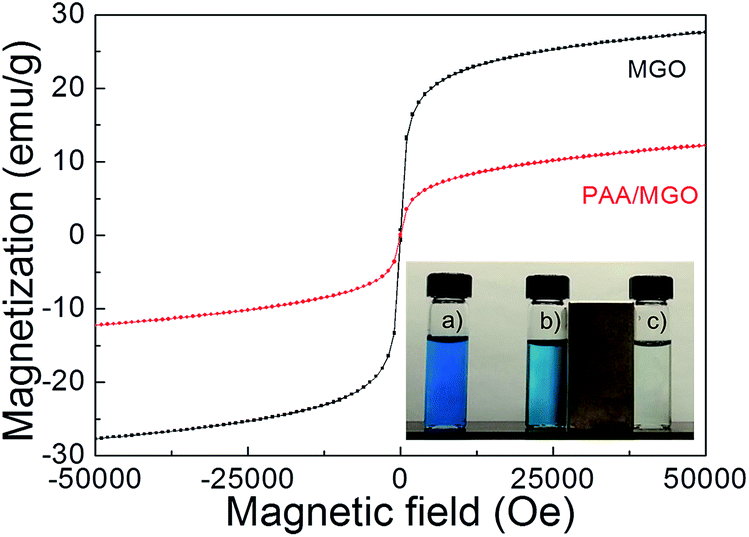
Fig. 7 shows the magnetic responses of MGO and PAA/MGO composites, both of which show typical superparamagnetic behavior. The saturation magnetizations of MGO and PAA/MGO are 27.7 emu g−1 and 12.3 emu g−1, respectively. The reduced saturation magnetization of PAA/MGO as compared to MGO could be attributed to the less mass fraction of magnetic component in the composite. The inset in Fig. 7 shows a picture of the mixtures (pH = 7) of 20 mg L−1 MB solutions (5 mL) with (a) 0.5 mL of DI water, (b) 0.5 mL of MGO (2 mg) solution and (c) 0.5 mL of PAA/MGO (2 mg) solution. Under an external magnetic field, both MGO and PAA/MGO can be easily separated, but only the residue solution for the PAA/MGO case can be totally discolored (in ∼2 minutes), which demonstrates the magnetism and different adsorption capacity of the nanocomposite adsorbents.
 |
| | Fig. 7 Magnetic hysteresis loops of MGO and PAA/MGO. The inset shows the mixtures of 20 mg L−1 MB solutions (5 mL) and (a) 0.5 mL of DI water, (b) 0.5 mL of MGO (2 mg) solution and (c) 0.5 mL of PAA/MGO (2 mg) solution. | |
3.3. Adsorption tests
The adsorption capacity of MB on MGO and PAA/MGO composites was investigated by batch tests. Fig. 8 shows the adsorption capacity qt of MB on MGO and PAA/MGO as a function of adsorption time t. Fig. 8 exhibits that the adsorption of MB drastically increases with time in the first few minutes, and then gradually reaches equilibrium. The adsorption capacity qt at time t of MB on PAA/MGO was much higher than that on MGO. For example, qt = 100 mg g−1 and 37 mg g−1 at t = 20 min for PAA/MGO and MGO, respectively, showing that the functionalization of PAA on MGO significantly improved the adsorption capacity of MB of the composite materials.
 |
| | Fig. 8 Adsorption capacity qt of MB on MGO and PAA/MGO as a function of adsorption time t, with 20 mg L−1 initial concentration of MB, pH = 7. Inset shows the adsorption profile of MB on MGO and PAA/MGO in the initial adsorption stage. | |
The pseudo-first-order and pseudo-second-order kinetic models were used to study the adsorption kinetics of MB on MGO and PAA/MGO.69,70 The pseudo-first-order kinetic model assumes that the adsorption rate is proportional to the available number of adsorption sites; while the pseudo-second-order kinetic model assumes that the adsorption rate is proportional to the square of available number of adsorption sites.51
Pseudo-first-order kinetic model:
| |
 | (3) |
Pseudo-second-order kinetic model:
| |
 | (4) |
qe and
qt (mg g
−1) represent the amounts of MB adsorbed at equilibrium and at any adsorption time
t.
k1 (min
−1) and
k2 (g mg
−1 min
−1) are pseudo-first and pseudo-second order rate constants, respectively.
Fig. 9a and b show the fitting for the adsorption of MB on MGO and PAA/MGO using the pseudo-first-order kinetics and pseudo-second-order kinetics, respectively. All kinetic parameters obtained by linear regression of the two kinetic models are summarized in
Table 2. The higher correlation coefficient values (
R2) in
Table 2 and the good agreement between the measured
qe,exp and the calculated
qe,cal indicate a better fitting of the data using the pseudo-second-order kinetic model than the pseudo-first-order model for the adsorption behaviors of MB on MGO and PAA/MGO.
 |
| | Fig. 9 Fitting of the adsorption kinetics of MB on MGO and PAA/MGO using (a) the pseudo-first-order kinetic model and (b) pseudo-second-order kinetic model. Symbols are experimental values and solid lines are the fittings using the two kinetic models. | |
Table 2 Parameters for the fitting of the adsorption of MB on MGO and PAA/MGO using the pseudo-first-order and pseudo-second-order kinetic models
| Adsorbents |
qe,exp (mg g−1) |
Pseudo-second-order kinetics |
Pseudo-second-order kinetics |
| k1 (min−1) |
qe,cal (mg g−1) |
R2 |
k2 (g mg−1 min−1) |
qe,cal (mg g−1) |
R2 |
| MGO |
61.2 |
0.0027 |
19.26 |
0.58862 |
0.0023 |
60.9 |
0.99687 |
| PAA/MGO |
117.5 |
0.0030 |
19.26 |
0.91559 |
0.0007 |
118.3 |
0.99998 |
To better understand the interactions between the adsorbents and MB, the adsorption data has been analyzed using Langmuir and Freundlich isotherm models, as shown in eqn (5) and (6), respectively.71,72 Langmuir model assumes monolayer adsorption of adsorbate on a homogeneous surface of adsorbent. Ce (mg L−1) is the equilibrium concentration of the MB. qm (mg g−1) is the maximum Langmuir monolayer adsorption capacity. b is related to the energy of the adsorption.73 qm and b can be obtained from the fitting of experimental data by eqn (5). The Freundlich isotherm model is an empirical model assuming a heterogeneous surface of adsorbent. Kf and n are the indicators of adsorption capacity and adsorption intensity, respectively, which can be determined from the fitting of experimental data by eqn (6).
Langmuir isotherm equation:
| |
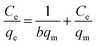 | (5) |
Freundlich isotherm equation:
| |
 | (6) |
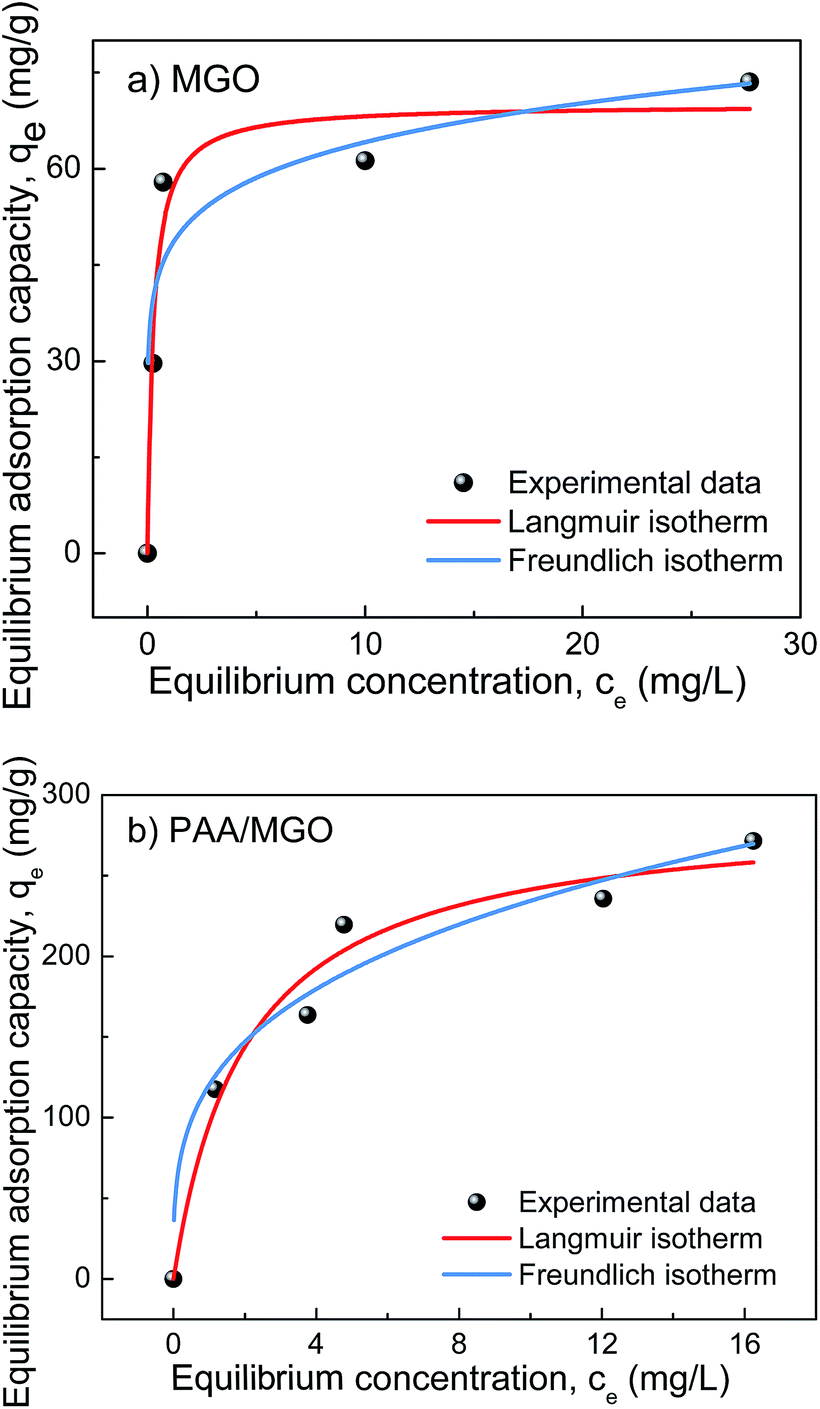
Fig. 10a and b show the fitting of adsorption data using the Langmuir and Freundlich models for the adsorption of MB on MGO and PAA/MGO, respectively and the fitting parameters are summarized in Table 3. The regression coefficients (R2) suggest that the Langmuir model fits the adsorption better, which implies that both MGO and PAA/MGO likely possess homogeneous adsorption surface and the adsorption of MB on these two adsorbents is likely monolayer. However, it should be noted previous studies have shown that certain heterogeneous materials/surfaces and heterogeneous adsorption could also obey the Langmuir model.71,74 The qm of MB on PAA/MGO (290.7 mg g−1) is much higher than qm of MB on MGO (70.0 mg g−1) due to the incorporation of PAA. At pH 7 for the adsorption tests, the deprotonated carboxylic groups of PAA grafted on MGO can have electrostatic attraction with cationic MB. The hydrophilicity of PAA also facilitates the dispersion of PAA/MGO composite and enhances the stability of the suspension, facilitating the exposure of adsorbent surfaces to contact with MB molecules.
 |
| | Fig. 10 Isotherms of the adsorption of MB on (a) MGO and (b) PAA/MGO, with a contact time of 24 h. | |
Table 3 Fitting parameters of the adsorption of MB on MGO and PAA/MGO using Langmuir and Freundlich isotherm models
| |
Langmuir model |
Freundlich model |
| b (mg L−1) |
qm (mg g−1) |
R2 |
Kf |
n |
R2 |
| MGO |
3.80 |
70.0 |
0.95 |
47.48 |
7.66 |
0.62 |
| PAA/MGO |
0.49 |
290.7 |
0.97 |
120.24 |
3.45 |
0.88 |
3.4. Effects of pH
The effects of pH on zeta potential of MGO and PAA/MGO and the adsorption capacity, q, of MB on MGO and PAA/MGO were also investigated and the results are shown in Fig. 11. Fig. 11a shows that with increasing pH = 3 to 11 the zeta potential of both MGO and PAA/MGO decreases, and the zeta potential of PAA/MGO is lower than that of MGO over the whole pH range. The decreased zeta potential can be attributed to that the carboxylic groups on MGO and on the PAA chains of PAA/MGO deprotonate as pH increases leading to more negatively charged surfaces. The lower zeta potential of PAA/MGO compared with MGO is due to its higher content of carboxylic groups. Fig. 11b shows that adsorption capacity of MB on MGO increases with increasing pH over the whole range (pH = 3 to 11); while adsorption capacity of MB on PAA/MGO increases from pH = 3 to 7 achieving almost complete adsorption from pH = 7 to 11 (viz. PAA/MGO removed almost all the dye in the solutions). It is noted that PAA/MGO exhibits much higher adsorption capacity of MB than MGO over the whole pH range investigated. As MB molecule contains chloridion that can deionize in aqueous solution and carry positive charge, MB shows electrostatic attraction with negatively charged carboxylic groups.75 Therefore more negative zeta potential of MGO and PAA/MGO leads to higher adsorption capacity of MB. It is noted that the zeta potential of MGO at pH = 3 is ∼−6.1 mV and is almost neutral with negligible electrostatic attraction with MB. The limited adsorption capacity of MB on MGO and PAA/MGO even at pH 3 indicates that π–π interaction between GO surface and MB also plays a role in the adsorption process.
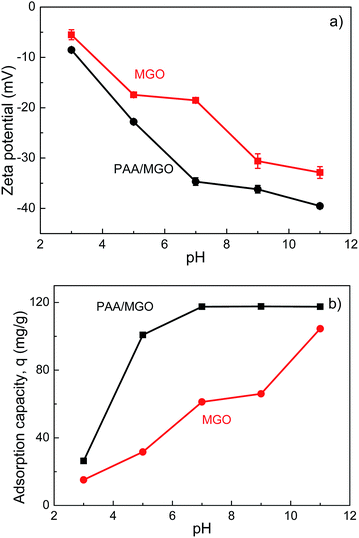 |
| | Fig. 11 (a) Zeta potential of MGO and PAA/MGO and (b) adsorption capacity of MB on MGO and PAA/MGO under various pH conditions. | |
3.5. Recyclable usage tests
The recyclability of adsorbents is an important factor for their practical application in water treatment. Adsorption–desorption cycles were carried out to test the recyclable usage of MGO and PAA/MGO on the removal of MB. Fig. 12 shows that after 5 cycles, the removal percentage of MB by PAA/MGO still remains >98%, while the removal percentage of MB by MGO drops gradually to about 31%. The better recyclability of PAA/MGO is likely due to the better dispersity of the adsorbents owing to the functionalization of hydrophilic PAA on MGO.
 |
| | Fig. 12 Removal percentages p% of MB by MGO and PAA/MGO for 5 cycles. | |
4. Conclusions
In this work, we report a facile method for the synthesis of polyacrylic acid functionalized magnetic Fe3O4 nanoparticle-graphene oxide nanocomposite (PAA/MGO). The structure and surface properties of MGO and PAA/MGO composites were characterized by various techniques including FTIR, XPS, TEM, HRTEM, TGA and zeta potential determinations. The adsorption behaviors of a model dye pollutant, methylene blue, on MGO and PAA/MGO were evaluated in batch tests. At pH 7, MGO and PAA/MGO nanocomposites show a maximum adsorption capacity of MB ∼70 mg g−1 and ∼291 mg g−1, respectively. The higher adsorption capacity of MB on PAA/MGO compared to MGO was attributed to the enhanced electrostatic attraction between positively charged MB molecules and negatively charged nanocomposite surfaces due to the higher content of deprotonated carboxyl groups on PAA/MGO. The removal capacity of MB by both MGO and PAA/MGO adsorbents also increases with increasing solution pH, and the zeta potential of both MGO and PAA/MGO decreases with increasing the solution pH which enhances the electrostatic attraction between MB and the composite surfaces. At pH 3, MGO and PAA/MGO become almost neutral with zeta potential close to 0 mV, both of which still show limited adsorption capacity to MB, indicating that π–π interaction between GO surface and MB also contributes to the adsorption process. Our results show that the PAA/MGO possesses rapid adsorption rate and higher adsorption capacity of MB than previously reported magnetic graphene or magnetic graphene oxide composites (∼44 to ∼190 mg g−1) under similar experimental conditions.33,76,77 PAA/MGO also can be easily separated under an external magnetic field with excellent recyclability and reusability. The PAA/MGO nanocomposite has great potential application as a novel adsorbent for the removal of cationic organic pollutants from wastewater.
Acknowledgements
We gratefully acknowledge the financial support from the Natural Resources Canada ecoENERGY Innovation Initiative (ecoEII), Natural Sciences and Engineering Research Council of Canada (NSERC), and Canadian Centre for Clean Coal/Carbon and Mineral Processing Technologies (C5MPT). We also acknowledge the support to some of the facilities used in this work from NSERC, the Canada Foundation for Innovation (CFI) and the Alberta Advanced Education & Technology Small Equipment Grants Program (AET/SEGP).
Notes and references
- S. Zhu, S. Jiao, Z. Liu, G. Pang and S. Feng, Environ. Sci.: Nano, 2014, 1, 172–180 RSC.
- G. Crini, Bioresour. Technol., 2006, 97, 1061–1085 CrossRef CAS PubMed.
- A. Ozcan, A. S. Ozcan and O. Gok, Adsorption kinetics and isotherms of anionic dye of reactive blue 19 from aqueous solutions onto DTMA-sepiolite, 2007 Search PubMed.
- A. L. Prasad and T. Santhi, Sustainable Environ. Res., 2012, 22, 113–122 CAS.
- V. K. Garg, R. Gupta, A. Bala Yadav and R. Kumar, Bioresour. Technol., 2003, 89, 121–124 CrossRef CAS.
- B. Shi, G. Li, D. Wang, C. Feng and H. Tang, J. Hazard. Mater., 2007, 143, 567–574 CrossRef CAS PubMed.
- E. Guibal and J. Roussy, React. Funct. Polym., 2007, 67, 33–42 CrossRef CAS PubMed.
- H. Lachheb, E. Puzenat, A. Houas, M. Ksibi, E. Elaloui, C. Guillard and J.-M. Herrmann, Appl. Catal., B, 2002, 39, 75–90 CrossRef CAS.
- D. Zhao, G. Sheng, C. Chen and X. Wang, Appl. Catal., B, 2012, 111, 303–308 CrossRef PubMed.
- N. Zhang, Y. Zhang and Y.-J. Xu, Nanoscale, 2012, 4, 5792–5813 RSC.
- M. Purkait, S. DasGupta and S. De, Sep. Purif. Technol., 2004, 37, 81–92 CrossRef CAS PubMed.
- N. Zaghbani, A. Hafiane and M. Dhahbi, Sep. Purif. Technol., 2007, 55, 117–124 CrossRef CAS PubMed.
- X. Zhuang, Y. Wan, C. Feng, Y. Shen and D. Zhao, Chem. Mater., 2009, 21, 706–716 CrossRef CAS.
- C. Wang, C. Feng, Y. Gao, X. Ma, Q. Wu and Z. Wang, Chem. Eng. J., 2011, 173, 92–97 CrossRef CAS PubMed.
- M. Iram, C. Guo, Y. Guan, A. Ishfaq and H. Liu, J. Hazard. Mater., 2010, 181, 1039–1050 CrossRef CAS PubMed.
- V. K. Gupta, J. Environ. Manage., 2009, 90, 2313–2342 CrossRef CAS PubMed.
- L. Li, L. Fan, C. Luo, H. Duan and X. Wang, RSC Adv., 2014, 4, 24679–24685 RSC.
- Q. Li, Q.-Y. Yue, H.-J. Sun, Y. Su and B.-Y. Gao, J. Environ. Manage., 2010, 91, 1601–1611 CrossRef CAS PubMed.
- S. K. Alpat, Ö. Özbayrak, Ş. Alpat and H. Akçay, J. Hazard. Mater., 2008, 151, 213–220 CrossRef CAS PubMed.
- B. Hameed, A. M. Din and A. Ahmad, J. Hazard. Mater., 2007, 141, 819–825 CrossRef CAS PubMed.
- G. Ramesha, A. Vijaya Kumara, H. Muralidhara and S. Sampath, J. Colloid Interface Sci., 2011, 361, 270–277 CrossRef CAS PubMed.
- C. E. Hamilton,Functionalization, Coordination, and Coating of Carbon Nanomaterials,PhD Thesis,University of Rice, 2009.
- G. Zhao, T. Wen, C. Chen and X. Wang, RSC Adv., 2012, 2, 9286–9303 RSC.
- H. Wang, X. Yuan, Y. Wu, H. Huang, X. Peng, G. Zeng, H. Zhong, J. Liang and M. Ren, Adv. Colloid Interface Sci., 2013, 195, 19–40 CrossRef PubMed.
- S. Wang, H. Sun, H. M. Ang and M. O. Tadé, Chem. Eng. J., 2013, 226, 336–347 CrossRef CAS PubMed.
- G. Zhao, L. Jiang, Y. He, J. Li, H. Dong, X. Wang and W. Hu, Adv. Mater., 2011, 23, 3959–3963 CrossRef CAS PubMed.
- W. Zhang, C. Zhou, W. Zhou, A. Lei, Q. Zhang, Q. Wan and B. Zou, Bull. Environ. Contam. Toxicol., 2011, 87, 86–90 CrossRef CAS PubMed.
- S. Stankovich, R. D. Piner, X. Chen, N. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- N. A. Travlou, G. Z. Kyzas, N. K. Lazaridis and E. A. Deliyanni, Langmuir, 2013, 29, 1657–1668 CrossRef CAS PubMed.
- S.-T. Yang, S. Chen, Y. Chang, A. Cao, Y. Liu and H. Wang, J. Colloid Interface Sci., 2011, 359, 24–29 CrossRef CAS PubMed.
- C. J. Madadrang, H. Y. Kim, G. Gao, N. Wang, J. Zhu, H. Feng, M. Gorring, M. L. Kasner and S. Hou, ACS Appl. Mater. Interfaces, 2012, 4, 1186–1193 CAS.
- L. Ai, C. Zhang and Z. Chen, J. Hazard. Mater., 2011, 192, 1515–1524 CrossRef CAS PubMed.
- G. Xie, P. Xi, H. Liu, F. Chen, L. Huang, Y. Shi, F. Hou, Z. Zeng, C. Shao and J. Wang, J. Mater. Chem., 2012, 22, 1033 RSC.
- F. He, J. Fan, D. Ma, L. Zhang, C. Leung and H. L. Chan, Carbon, 2010, 48, 3139–3144 CrossRef CAS PubMed.
- K. Meral and Ö. Metin, Turk. J. Chem., 2014, 38, 775–782 CrossRef CAS PubMed.
- S.-H. Huang and D.-H. Chen, J. Hazard. Mater., 2009, 163, 174–179 CrossRef CAS PubMed.
- L. Kan, Z. Xu and C. Gao, Macromolecules, 2010, 44, 444–452 CrossRef.
- Y. Yang, Y. Xie, L. Pang, M. Li, X. Song, J. Wen and H. Zhao, Langmuir, 2013, 29, 10727–10736 CrossRef CAS PubMed.
- J. Liu, H. Cao, J. Xiong and Z. Cheng, CrystEngComm, 2012, 14, 5140–5144 RSC.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I.-C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed.
- H. Shi, W. Li, L. Zhong and C. Xu, Ind. Eng. Chem. Res., 2014, 53, 1108–1118 CrossRef CAS.
- L. Li, L. Fan, C. Luo, H. Duan and X. Wang, RSC Adv., 2014, 4, 24679 RSC.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- A. Durdureanu-Angheluta, R. Ardeleanu, M. Pinteala, V. Harabagiu, H. Chiriac and B. Simionescu, Digest Journal of Nanomaterials and Biostructures, 2008, 3, 33–40 Search PubMed.
- F. Everaerts, M. Torrianni, M. Hendriks and J. Feijen, J. Biomed. Mater. Res., Part A, 2007, 85, 547–555 Search PubMed.
- M.-H. Liao and D.-H. Chen, J. Mater. Chem., 2002, 12, 3654–3659 RSC.
- S.-Y. Mak and D.-H. Chen, Dyes Pigm., 2004, 61, 93–98 CrossRef CAS PubMed.
- Y. Yao, S. Miao, S. Yu, L. P. Ma, H. Sun and S. Wang, J. Colloid Interface Sci., 2012, 379, 20–26 CrossRef CAS PubMed.
- Y. Yao, S. Miao, S. Liu, L. P. Ma, H. Sun and S. Wang, Chem. Eng. J., 2012, 184, 326–332 CrossRef CAS PubMed.
- L. Li, X. L. Liu, M. Gao, W. Hong, G. Z. Liu, L. Fan, B. Hu, Q. H. Xia, L. Liu, G. W. Song and Z. S. Xu, J. Mater. Chem. A, 2014, 2, 1795 CAS.
- S.-M. Paek, E. Yoo and I. Honma, Nano Lett., 2009, 9, 72–75 CrossRef CAS PubMed.
- G. K. Ramesha, A. V. Kumara, H. B. Muralidhara and S. Sampath, J. Colloid Interface Sci., 2011, 361, 270–277 CrossRef CAS PubMed.
- Y. Xie, B. Yan, H. Xu, J. Chen, Q. Liu, Y. Deng and H. Zeng, ACS Appl. Mater. Interfaces, 2014, 6, 8845–8852 CAS.
- Y. Fu, J. Wang, Q. Liu and H. Zeng, Carbon, 2014, 77, 710–721 CrossRef CAS PubMed.
- G. Wang, S. Yang, Z. Wei, X. Dong, H. Wang and M. Qi, Polym. Bull., 2013, 70, 2359–2371 CrossRef CAS PubMed.
- Z. Yang, X.-H. Chen, S.-Z. Xia, Y.-X. Pu, H.-Y. Xu, W.-H. Li, L.-S. Xu, B. Yi and W.-Y. Pan, J. Mater. Sci., 2007, 42, 9447–9452 CrossRef CAS PubMed.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- Y. Dong, M. Hu, R. Ma, H. Cheng, S. Yang, Y. Y. Li and J. A. Zapien, CrystEngComm, 2013, 15, 1324–1331 RSC.
- Y. Jin, C. Jia, S.-W. Huang, M. O'Donnell and X. Gao, Nat. Commun., 2010, 1, 41 CrossRef PubMed.
- L. Reimer and H. Kohl, Transmission electron microscopy: physics of image formation, Springer Science & Business Media, 2008 Search PubMed.
- P. Gentsch, H. Gilde and L. Reimer, J. Microsc., 1974, 100, 81–92 CrossRef PubMed.
- G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts, John Wiley & Sons, 2004 Search PubMed.
- P. Larkin, Infrared and Raman spectroscopy; principles and spectral interpretation, Elsevier, 2011 Search PubMed.
- T. Shimanouchi, Tables of Molecular Vibrational Frequencies Consolidated, DTIC Document, 1972, vol. 1 Search PubMed.
- S. Some, Y. Kim, Y. Yoon, H. Yoo, S. Lee, Y. Park and H. Lee, Sci. Rep., 2013, 3, 1929 Search PubMed.
- Y. Fu, Water-Dispersible Magnetic Particle–Graphene Oxide Composites: Synthesis, Characterization and Application in the Removal of Selenium, Master Thesis, University of Alberta, 2014.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. A. Connors, Chemical kinetics: the study of reaction rates in solution, John Wiley & Sons, 1990 Search PubMed.
- J. Toth, Adsorption, CRC Press, 2002 Search PubMed.
- I. Langmuir, J. Am. Chem. Soc., 1916, 38, 2221–2295 CrossRef CAS.
- H. Freundlich, Zeitschrift für Physikalische, 1906, 57, 384–470 Search PubMed.
- R. I. Masel, Principles of adsorption and reaction on solid surfaces, John Wiley & Sons, 1996 Search PubMed.
- A. Dąbrowski, Adv. Colloid Interface Sci., 2001, 93, 135–224 CrossRef.
- E. M. Tuite and J. M. Kelly, J. Photochem. Photobiol., B, 1993, 21, 103–124 CrossRef CAS.
- F. He, J. Fan, D. Ma, L. Zhang, C. Leung and H. L. Chan, Carbon, 2010, 48, 3139–3144 CrossRef CAS PubMed.
- L. Li, X. L. Liu, M. Gao, W. Hong, G. Z. Liu, L. Fan, B. Hu, Q. H. Xia, L. Liu and G. W. Song, J. Mater. Chem. A, 2014, 2, 1795–1801 CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.