
Curcumin-loaded, folic acid-functionalized magnetite particles for targeted drug delivery†
Abstract
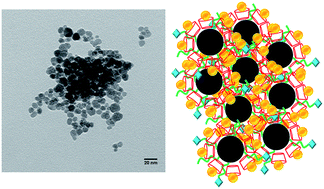
Precipitation of Fe3O4 from FeSO4 in the presence of NH4OH, amine-terminated poly(propylene glycol) (PPG-NH2) and β-cyclodextrin (β-CD) afforded magnetite particles coated with PPG-NH2 and β-CD. The sizes of the particles, as characterized by dynamic light scattering and electron microscopy, depended on the preparation conditions, but the essential magnetite structure and magnetic properties were retained in the presence of the surface coating. The terminal amine of PPG-NH2 was used for the conjugation of folic acid (FA), with the aim of targeting cancer cells overexpressing the FA receptor. Curcumin, a promising chemotherapeutic exhibiting low aqueous solubility and rapid metabolic degradation was encapsulated into the β-CD cavity to enhance its delivery to cancer cells. The loading of curcumin and its release rate was found to depend on the particle size and composition, with sustained curcumin release observed for some systems. The uptake of the particles into MDA-MB-231 and MDA-MB-468 human breast cancer cells was compared and the toxicities of the curcumin-loaded particles were compared to the free drug and to particles without curcumin.
 Please wait while we load your content...
Please wait while we load your content...

