
NCN pincer palladium complexes based on 1,3-dipicolyl-3,4,5,6-tetrahydropyrimidin-2-ylidenes: synthesis, characterization and catalytic activities†
Liangru Yang*,
Xinchi Zhang,
Pu Mao*,
Yongmei Xiao,
Huanyu Bian,
Jinwei Yuan,
Wenpeng Mai and
Lingbo Qu
School of Chemistry and Chemical Engineering, Henan University of Technology, Zhengzhou 450001, P. R. China. E-mail: lryang@haut.edu.cn; pumao@haut.edu.cn
First published on 5th March 2015
Abstract
The synthesis of novel pincer palladium complexes containing ring expanded six-membered N-heterocyclic carbenes (NHCs) via direct metallation of the precursors tetrahydropyrimidin-1-ium hexafluorophosphates in the presence of NaN(SiMe3)2 is presented. The structure has been characterized unambiguously by X-ray single crystal analysis. Catalytic activity investigation showed that the complexes catalyzed the Heck reaction of aryl bromides with acrylate/styrene efficiently when using Et3N as base and DMA as solvent.
Introduction
Since the successful isolation and characterization of the first stable N-heterocyclic carbene (NHC) by Arduengo et al. in 1991,1 these molecules have been widely used as ancillary ligands for the preparation of transition-metal-based catalysts.2 Today, NHC metal complexes rank among the most powerful tools in organic chemistry, with numerous publications related to their coordination chemistry and catalytic properties being reported.3 NHC palladium complexes are attracting a considerable amount of interest because of their easy accessibility, high thermal stability, and remarkable catalytic activities in various C–C coupling reactions. Numerous functionalized NHC palladium complexes have been synthesized and applied to catalytic hydrogenation, hydrophosphination, C–H functionalization, Suzuki–Miyaura reactions, Mizoroki–Heck reactions, Buchwald–Hartwig reactions, etc. successfully.4 However, most of the reported NHC ligands are based on five-membered heterocyclic rings (imidazol-2-ylidenes, imidazolin-2-ylidenes, or benzimidazolin-2-ylidene, etc.).In recently years, the ring expanded NHCs based on six-, seven-, or eight-membered heterocyclic rings began to attract extensive attention due to the enhanced σ-donor ability and easy tunability of the electronic property and steric effect of the ligands.5 Various kinds of ring expanded NHC metal complexes, including Ag,6 Au(I),7 Pd,8 Ni,9 Cu,10 Ru,11 Rh,12 and Ir,13 etc. have been synthesized, characterized, and applied to organic transformations successfully. For example, the ring expanded six-, or seven-membered NHC copper(I) complexes have been proved to be effective catalysts for the 1,3-dipolar cycloaddition of alkynes and azide.10a A ring-expanded six-membered NHC Nickel(I) complex has been reported to be a useful precursor for catalytic hydrodehalogenation.9 Palladium(II) complexes bearing ring expanded NHCs have been demonstrated to be effective catalysts for the intramolecular aerobic oxidative amination of alkenes,8a the Heck reaction of aliphatic and aromatic vinyl compounds with aryl bromides and chlorides,8c Suzuki–Miyaura cross-coupling of aryl bromide and chloride, and the catalytic dehalogenation of aryl chloride.5a
The pincer architecture, which provides a preorganised backbone featuring unique properties of high stability and modular variability has also been extensively studied in a wide range of fields.14 Therefore, the inclusion of NHC donors within pincer systems has attracted an increasing level of interest, with a particular emphasis on their catalytic potential.15 Multiple pincer-type NHC palladium complexes carrying different donor moieties have been prepared and employed as catalysts for a number of catalytic organic transformations (Fig. 1).16 For example, the palladium complexes of phosphine/NHC-based pincer ligand PCNHCP (B) have been reported to be active catalysts for Suzuki coupling and Heck coupling reactions.16b,16c All these systems, however, contain NHC scaffolds based on five-membered heterocyclic rings.
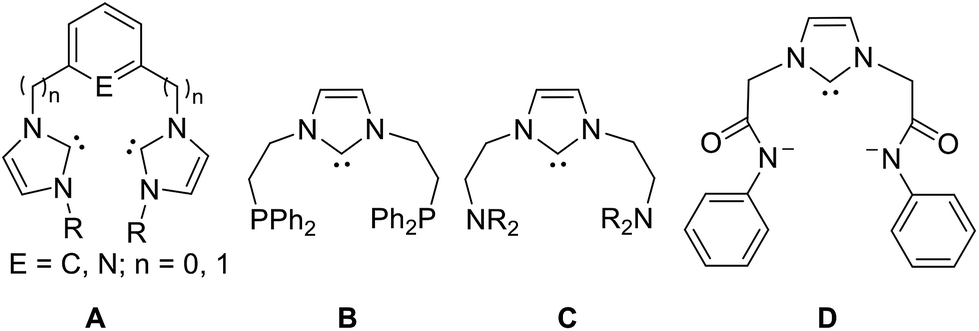 | ||
| Fig. 1 NHC-based pincer ligands. | ||
Based on the reports mentioned above, it would be of interest to explore whether the introduction of ring expanded NHCs to a pincer framework will result in new pincer complexes displaying novel reactivity and enhanced catalytic activities. While to the best of our knowledge, the only report on the pincer ring expanded NHC complexes was about the phosphine functionalized dihydroperimidine-based NHC Rh, Ir complexes generated via chelate-assisted double C–H activation of substituted 2,3-dihydroperimidine proligands.17 In view of the strong donating properties of the ring expanded NHCs and obvious rare of precedent studies on their pincer metal complexes, here we report the synthesis, characterization and catalytic activity of picolyl functionalized pincer six-membered NHC palladium complexes based on tetrahydropyrimidin-2-ylidenes. As expected, the pincer six-membered NHC palladium complexes proved remarkably stable toward air and moisture, and showed high catalytic activity toward Heck reaction. These results underline the high potential of this class of carbene ligands in catalysis.
Results and discussion
Synthesis of tetrahydropyrimidin-1-ium derivatives
In literature report, dehydrogenation of hexahydropyrimidine by NBS have been used quite often to synthesis tetrahydropyrimidin-1-iums salts, along with other procedures, including quarterisation of tetrahydropyrimidine, and direct cyclization of N,N′-dialkylpropan-1,3-diamines with methyl orthoformate catalyzed by acid.18 It features the obvious advantages of high yield and repeatability, and easy purification of the product. Here we synthesized the pincer six-membered NHC precursors 1,3-dipicolyl-3,4,5,6-tetrahydropyrimidin-1-ium salts through the dehydrogenation of 1,3-disubstituted hexahydropyrimidine by NBS. The synthetic procedure is shown in Scheme 1. Condensation of 1,3-propandiamines with pyridine-2-formaldehyde in methanol produced Schiff bases in high yields. Reduction of the resulting Schiff bases with NaBH4 lead to the formation of N,N′-dialkylpropan-1,3-diamines. The following reaction with aqueous formaldehyde in methanol afforded the 1,3-dipicolyl-hexahydropyrimidines (1a–b), which were then treated by NBS to obtain the tetrahydropyridin-1-ium bromides (2a–b). Anion exchange with NH4PF6 in ethanol/H2O produced the corresponding tetrahydropyridin-1-ium hexafluorophosphates (3a–b). The ligands 3a–b were obtained analytically pure in high yields after recrystallization. Their structures were fully characterized by various NMR techniques and mass spectra measurement, and in the case of complex 3b, by X-ray crystallographic determination. In hexahydropyrimidines 1a–b, the proton resonances of the methylene groups on picolyl occurred as singlet signals at 3.61 and 3.51, respectively. The resonances of NCH2N protons appeared at 3.19 and 3.06 ppm, respectively. While in the corresponding tetrahydropyridin-1-ium salts (2a–b, 3a–b), the proton resonances of the methylene groups on picolyl downfielded within the range of 4.86–4.84 ppm, and the resonances of NCHN protons downfielded within the range of 9.07–8.91 ppm, respectively. Single crystals of 3b suitable for X-ray diffraction analysis were obtained by slow diffusion of diethyl ether to a dichloromethane solution of 3b.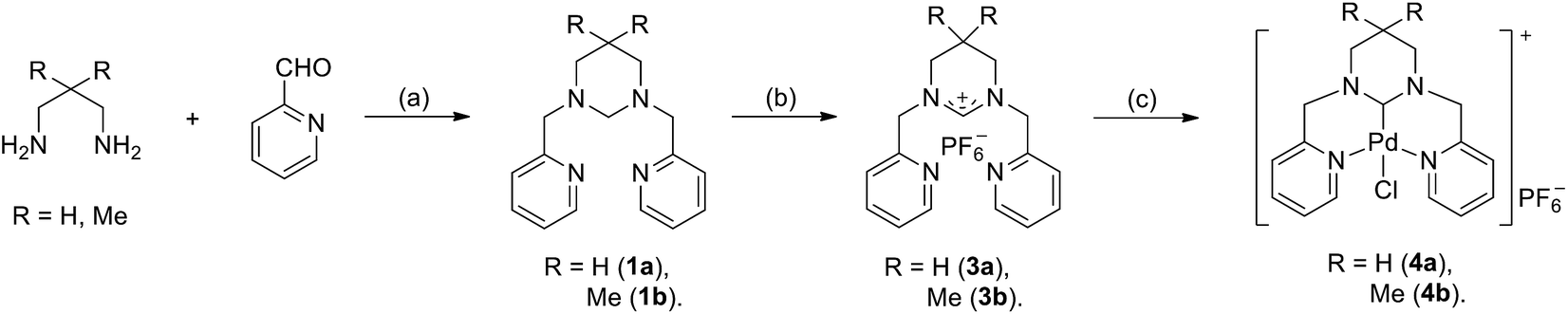 | ||
| Scheme 1 Synthesis of the ligands and pincer ring expanded NHC–Pd complexes. (a) (i) MeOH; (ii) NaBH4, MeOH, 0–70 °C; (iii) HCHO, MeOH; (b) (i) NBS, DME; (ii) NH4PF6, EtOH/H2O; (c) PdCl2, NaN(SiMe3)2, pyridine, 140 °C. | ||
Fig. 2 shows the molecular structure of 3b with selected bond lengths and bond angles listed in the caption. The bonding within the pyrimidinyl ring indicates a pattern of delocalization that extends from N(1) to N(2) through C(1), with N(1)–C(1) [1.305(3) Å] and N(2)–C(1) [1.305(4) Å] being significantly shorter than those between N(1)–C(2) [1.463(4) Å] and N(2)–C(4) [1.465(4) Å]. The nitrogen donors of the picolyl groups are rotated away from C(1), showing that the donor arms rotating freely to take up positions suitable for chelation to a metal ligated at C(1).
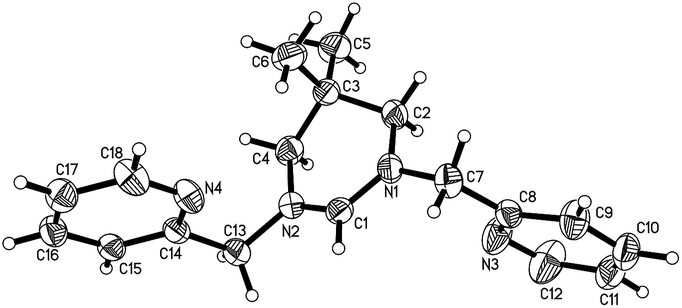 | ||
| Fig. 2 Molecular structure of 3b (counter ion omitted, 50% displacement ellipsoids). Selected bond lengths (Å) and angles (deg): N(1)–C(1) 1.305(3), N(1)–C(2) 1.463(4), N(1)–C(7) 1.458(4), N(2)–C(1) 1.305(4), N(2)–C(4) 1.465(4), N(2)–C(13) 1.458(3), N(1)–C(1)–N(2) 124.1(2), C(1)–N(1)–C(2) 120.3(2), C(1)–N(2)–C(4) 121.3(2), C(1)–N(1)–C(7) 120.1(2), C(2)–N(1)–C(7) 119.2(2), C(1)–N(2)–C(13) 119.9 (2), C(4)–N(2)–C(13) 118.6 (2). | ||
Synthesis of pincer NHC palladium complexes
Following a similar literature report,19 the pincer NHC–Pd complexes (4a–b) were prepared by heating the corresponding tetrahydropyridin-1-ium hexafluorophosphates (3a or 3b) and PdCl2 in the presence of NaN(SiMe3)2 as a base in pyridine at 140 °C (Scheme 1). The formation of the pincer NHC palladium complexes was observed from the studies of NMR spectra, showing the conspicuous absence of the NCHN resonances of the reacting cationic tetrahydropyrimidin-1-ium hexafluorophosphates and the appearance of the highly downfield shifted at 174.9 (4a) and 174.0 (4b) ppm in the 13C NMR spectra, which should be attributed to the new Pd–Ccarbene resonance. In addition, the proton resonances of the methylene groups on picolyl occurred as a doublet of doublets at 5.30 and 4.80 (4a), and 5.38 and 4.78 (4b) ppm, respectively. This confirms the formation of the chelate ring, which makes the two protons of the methylene group on the picolyl magnetic unequal. At the same time, the pyridine proton resonances appeared within the ranges of 9.00–7.63 (4a) and 9.01–7.65 (4b) ppm, obviously downfielded compared to those of the tetrahydropyrimidin-1-ium hexafluorophosphates 3a (8.63–7.41 ppm) and 3b (8.94–7.41 ppm), supporting the coordination of pyridine to the palladium center. The complex 4b has been further characterized unambiguously by the single-crystal X-ray diffraction studies.Slow diffusion of diethyl ether to a concentrated acetonitrile solution of 4b produced single crystals suitable for X-ray diffraction analysis. The molecular structure is shown in Fig. 3, selected bond lengths and bond angles listed in the caption.
 | ||
| Fig. 3 Molecular structure of 4b (counter ion omitted, 50% displacement ellipsoids). Selected bond lengths (Å) and angles (deg): Pd(1)–C(1) 1.975(5), Pd(1)–N(3) 2.037(4), Pd(1)–N(4) 2.035(4), Pd(1)–Cl(1) 2.3619(14), N(1)–C(1) 1.334(7), N(1)–C(2) 1.452(7), N(1)–C(7) 1.468(7), N(2)–C(1) 1.316(7), N(2)–C(4) 1.467(8), N(2)–C(13) 1.453(7), C(1)–Pd(1)–Cl(1) 176.89(16), N(3)–Pd(1)–N(4) 175.83(18), C(1)–Pd(1)–N(3) 87.8(2), C(1)–Pd(1)–N(4) 88.08(19), N(3)–Pd(1)–Cl(1) 91.65(14), N(4)–Pd(1)–Cl(1) 92.51(13), N(1)–C(1)–N(2) 120.6(5), C(1)6N(1)–C(2) 122.0(5), C(1)–N(2)–C(4) 122.8(5), C(1)–N(1)–C(7) 120.1(2), C(2)–N(1)–C(7) 119.5(5), C(1)–N(2)–C(13) 119.0(5), C(4)–N(2)–C(13) 117.9(5). | ||
As shown in Fig. 3, the palladium atom in 4b adopts a slightly distorted square-planar coordination bonded to carbene and two pyridinyl nitrogen donors, with the two fused six-membered chelate rings of the ‘pincer’ both exist in boat conformation. As complex 4b represents the only example of a structurally characterized pincer six-membered NHC palladium complex that we are aware of, we compare the structure with pincer five-member NHC palladium complexes, and non-chelating six-membered NHC palladium complex, respectively. The Pd–Ccarbene bond distance of 1.975(5) Å is slightly longer than those found in the dipicolyl functionalized pincer NHC palladium bearing imidazol-2-ylidene (1.929(4) Å) or imidazolin-2-ylidene (1.936(2) Å),20 while very similar to those found in the pincer NHC palladium complexes bearing diphosphine-substituted benzimidazolin-2-ylidene (1.974(5) Å and 1.980(6) Å) or imidazol-2-ylidene (1.983(7) Å) ligands reported by Hahn and Lee,16b,c and non-chelating six-membered NHC palladium complex reported by Cavell (1.983(3) Å) and Ghosh (1.9812(19) Å).5a,19
The Pd–N distances (2.037(4) Å and 2.035(4) Å) and Pd–Cl distance (2.3619(14) Å), are very similar to those of the picolyl functionalized NHC palladium carrying imidazol-2-ylidene (2.073(3) Å and 2.062(3) Å, 2.3737(3) Å) or imidazolin-2-ylidene (2.066(2) Å and 2.054(4) Å, 2.3688(5) Å).20
Catalytic studies
Initially, running the Heck reaction of bromobenzene and n-butyl acrylate catalyzed by 1.0% complex 4a in DMA as a model reaction, a brief screening of the base and solvent was conducted (Table 1). Among the bases tested, organic base NEt3 afforded the moderate yield. Inorganic bases t-BuOK, K2CO3, Na2CO3, and Cs2CO3 all produced very low yields (Table 1, entries 1–5). Tests of different solvents proved DMA to be the proper solvent. Reaction in DMF and toluene produced the target product in low yields, and the reaction in DME or dioxane did not occur at all (Table 1, entries 6–9). Increasing the catalyst loading to 2.5% raised yield to 98%.
|
||||
|---|---|---|---|---|
| Entrya | Solvent | Base | Cat. (mol%) | Yieldb (%) |
| a Reaction condition: 0.5 mmol bromobenzene, 0.75 mmol n-butyl acrylate, 0.75 mmol base, 2 mL solvent, 135 °C, 12 h.b Yields determined by HPLC. | ||||
| 1 | DMA | Et3N | 1.0 | 54 |
| 2 | DMA | t-BuOK | 1.0 | <5 |
| 3 | DMA | K2CO3 | 1.0 | <5 |
| 4 | DMA | Na2CO3 | 1.0 | <5 |
| 5 | DMA | Cs2CO3 | 1.0 | <5 |
| 6 | DMF | Et3N | 1.0 | 40 |
| 7 | DME | Et3N | 1.0 | 0 |
| 8 | Toluene | Et3N | 1.0 | 36 |
| 9 | Dioxane | Et3N | 1.0 | 0 |
| 10 | DMA | Et3N | 1.5 | 70 |
| 11 | DMA | Et3N | 2.5 | 98 |
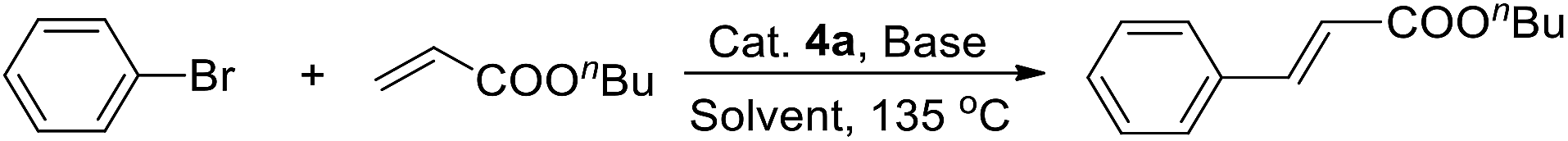
Under the standard conditions, using Et3N as base and DMA as solvent, the catalytic activity of complexes 4a and 4b towards Heck reaction of a series of aryl bromides with olefins (acrylate and styrene) were investigated and the results are summarized in Table 2. The results showed that complexes 4a and 4b presented almost equally catalytic efficiency, producing the target products in moderate to high yields. The substituents on the phenyl ring of the aryl bromides did not show any obvious electronic effect, while the steric effect was obvious. In some cases of ortho-substituted phenyl bromide and 1-naphthyl bromide, comparatively low yields were obtained (Table 2, entries 10–15, 23 and 24).
|
|||||
|---|---|---|---|---|---|
| Entrya | Ar–Br | Olefines | Products | Yieldsb (%) | |
| Cat. 4a | Cat. 4b | ||||
| a Reaction condition: 0.5 mmol aryl bromide 0.75 mmol olefine, 0.75 mmol Et3N, 0.0125 mmol 4a or 4b, 2 mL solvent, 135 °C, 12 h.b Isolated yields. | |||||
| 1 | C6H5Br | Methyl acrylate | 5a | 95 | 93 |
| 2 | C6H5Br | n-Butyl acrylate | 5b | 94 | 90 |
| 3 | C6H5Br | Styrene | 5c | 88 | 86 |
| 4 | 2-Me–C6H4–Br | Methyl acrylate | 5d | 89 | 88 |
| 5 | 2-Me–C6H4–Br | n-Butyl acrylate | 5e | 89 | 87 |
| 6 | 2-Me–C6H4–Br | Styrene | 5f | 76 | 80 |
| 7 | 4-Me–C6H4–Br | Methyl acrylate | 5g | 93 | 90 |
| 8 | 4-Me–C6H4–Br | n-Butyl acrylate | 5h | 91 | 89 |
| 9 | 4-Me–C6H4–Br | Styrene | 5i | 78 | 75 |
| 10 | 2,4-Me2–C6H3–Br | Methyl acrylate | 5j | 82 | 80 |
| 11 | 2,4-Me2–C6H3–Br | n-Butyl acrylate | 5k | 78 | 79 |
| 12 | 2,4-Me2–C6H3–Br | Styrene | 5l | 70 | 68 |
| 13 | 2-OCH3–C6H4–Br | Methyl acrylate | 5m | 81 | 83 |
| 14 | 2-OCH3–C6H4–Br | n-Butyl acrylate | 5n | 78 | 80 |
| 15 | 2-OCH3–C6H4–Br | Styrene | 5o | 68 | 72 |
| 16 | 4-OCH3–C6H4–Br | Methyl acrylate | 5p | 91 | 88 |
| 17 | 4-OCH3–C6H4–Br | n-Butyl acrylate | 5q | 88 | 85 |
| 18 | 4-OCH3–C6H4–Br | Styrene | 5r | 76 | 80 |
| 19 | 4-COCH3–C6H4–Br | Methyl acrylate | 5s | 95 | 94 |
| 20 | 4-COCH3–C6H4–Br | n-Butyl acrylate | 5t | 89 | 87 |
| 21 | 4-COCH3–C6H4–Br | Styrene | 5u | 79 | 76 |
| 22 | 1-C10H7–Br | Methyl acrylate | 5v | 87 | 84 |
| 23 | 1-C10H7–Br | n-Butyl acrylate | 5w | 65 | 60 |
| 24 | 1-C10H7–Br | Styrene | 5x | 70 | 68 |

The Heck reaction of phenyl bromide or para-substituted phenyl bromide with olefines was achieved in less time or lower catalyst loading, using pincer diphosphine-substituted imidazol-2-ylidene palladium complex,16c than the results reported here for complexes 4a and 4b. While for the Heck reaction of ortho-substituted phenyl bromide or 1-naphthyl bromide, moderate to good yields were obtained using complexes 4a and 4b. This suggests that there is considerable potential to improve the activity of these complexes, with the possibility of developing these complexes as catalysts for the Heck reaction of more substituted, sterically demanding substrates.
Experimental section
General consideration
All solvents and chemicals were used as received or dried with standard methods and freshly distilled prior to use if needed. NMR spectra were recorded at 25 °C on a 400 MHz Bruker spectrometer. Chemical shifts (δ in ppm, coupling constants J in Hz) were referenced to the residual solvent resonances. Elemental analyses were obtained from a thermo Flash 2000. ESI-MS spectra were recorded on a Bruker Esquire 3000.Synthesis of 1,3-dipicolyl hexahydropyrimidines (1a–b)
A methanol solution (50 mL) containing pyridine-2-formaldehyde (30 mmol, 3.21 g) and 1,3-propanediamine (15 mmol, 1.11 g) were stirred at room temperature for 5 h. Infrared detection showed the disappearance of the carbonyl group. Additional 20 mL methanol was added and the mixture was put into an ice-bath, then NaBH4 (120 mmol, 4.54 g) was added portion-wise for 1 h. The mixture was then warmed up to room temperature and then heated to 70 °C overnight. The solvent was then evaporated and the residue was poured into a mixture of water (40 mL) and CH2Cl2 (40 mL). The resulting suspension liquid was filtered and the filtrate was extracted by CH2Cl2 (20 mL) for 3 times. The combined organic phase was evaporated and the residue obtained was dissolved in methanol (10 mL) for the following reaction directly. The solution was then treated with aqueous HCHO solution (36.5%, 15 mmol). The mixture was stirred at room temperature for 6 h before being evaporated. Purification of the residue by flash chromatography (silica, acetone/CH2Cl2/Et3N = 2/8/1, v/v/v) afforded the pure products.Synthesis of 1,3-dipicolyl tetrahydropyrimidin-1-ium bromides (2a–b)
Hexahydropyrimidine (1a or 1b, 5 mmol) was dissolved in DME (20 mL). NBS (5 mmol, 0.89 g) was added portion-wise and the resulting mixture was stirred at room temperature for 3 h, during which time a white precipitate formed. The precipitate was filtered and washed with DME. Crystallization of the precipitate from CH2Cl2/diethyl ether produced the pure products.Synthesis of 1,3-dipicolyl tetrahydropyrimidin-1-ium hexafluorophosphates (3a–b)
NH4PF6 (3 mmol, 0.49 g) was dissolved in water (5 mL). The water solution was then combined with an ethanol (25 mL) solution of tetrahydropyrimidin-1-ium bromides (2a or 2b, 2 mmol) and the mixture was then stirred at room temperature for 5 h. The solvent was then evaporated and the residue was poured into a mixture of water (10 mL) and CH2Cl2 (10 mL). The resulting solution was then extracted by CH2Cl2 (10 mL) for 3 times. The combined organic phase was then washed by water, dried and evaporated. Crystallization of the resulting white powder from CH2Cl2/diethyl ether produced the pure products.Synthesis of 1,3-dipicolyl-3,4,5,6-tetrahydropyrimidin-2-ylidenes palladium complexes (4a–b)
A mixture of tetrahydropyrimidin-1-ium hexafluorophosphate (3a or 3b, 2.0 mmol), PdCl2 (2.0 mmol, 0.35 g) and NaN(SiMe3)2 (1.2 mL, 2.0 M in THF) in pyridine (6 mL) was heated at 140 °C for 12 h. The reaction mixture was then evaporated and purification of the residue by column chromatography (silica, CHCl3/diethyl ether, gradient elution) produced the pure palladium complexes.General procedure for the Heck reaction
The Heck reaction was conducted in a parallel reactor. In a typical reaction, A Schlenk tube was charged with aryl bromides (0.5 mmol), acrylate or styrene (0.75 mmol), base (0.75 mmol), pincer NHC–Pd complex 4a or 4b, and solvent (2 mL). The mixture was stirred at 135 °C for 12 h under Ar. After cooling, the reaction mixture was evaporated. Purification of the residue by flash chromatography on silica gel (hexanes/CH2Cl2 = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) afforded the pure products, which were characterized by 1H NMR and 13C NMR. The analytical data of the products were shown in the ESI.†
:1) afforded the pure products, which were characterized by 1H NMR and 13C NMR. The analytical data of the products were shown in the ESI.†
X-ray diffraction studies
The crystal data of 3b and 4b (ESI†) were collected on a Xcalibur, Eos, Gemini diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å). The crystals were kept at 291.15 K during data collection. Using Olex2,21 the structure was solved with the ShelXS22 structure solution program using Direct Methods and refined with the ShelXL22 refinement package using Least Squares minimization.Conclusions
Two novel NCN pincer ring expanded six-membered NHC palladium complexes based on 1,3-dipicolyl-3,4,5,6-tetrahydropyrimidin-2-ylidenes have been synthesized via the direct metallation of the corresponding tetrahydropyridin-1-ium salts. The structure were characterized unambiguously X-ray structure analysis, showing that the complex adopts a slightly distorted square-planar coordination with the two fused six-membered chelate rings of the ‘pincer’ both exist in boat conformation. Catalytic activity investigation showed that the complexes catalyzed the Heck reaction of aryl bromides with acrylate/styrene efficiently in DMA when using Et3N as base. The catalytic activity of this novel type of NHC complexes towards other organic transformation are currently in progress in our group.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (nos: 21172055 and 21302042), the Program for Innovative Research Team from Zhengzhou (131PCXTD605), and the Plan for Scientific Innovation Talent of Henan University of Technology (11CXRC10) for financial support to this work.Notes and references
- (a) A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef; (b) A. J. Arduengo III, M. Kline, J. C. Calabrese and F. Davidson, J. Am. Chem. Soc., 1991, 113, 9704 CrossRef.
- (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed; (b) J. C. Bernhammer, H. Singh and H. V. Huynh, Organometallics, 2014, 33, 4295 CrossRef CAS; (c) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 RSC; (d) O. Kühl, Functionalised N-Heterocyclic Carbene Complexes, Wiley: John Wiley & Sons Ltd, 2010 Search PubMed; (e) S. P. Nolan, N-heterocyclic carbenes in synthesis, Wiley-VCH, Weinheim, 2006 Search PubMed; (f) F. E. Hahn, Angew. Chem., Int. Ed., 2006, 45, 1348 CrossRef CAS PubMed; (g) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (h) W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed., 1997, 36, 2162 CrossRef CAS.
- (a) F. Wang, L. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804 CrossRef CAS PubMed; (b) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed; (c) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561 CrossRef CAS PubMed; (d) C. Samojłowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708 CrossRef PubMed; (e) D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef CAS PubMed.
- (a) L. Benhamou, C. Besnard and E. P. Kündig, Organometallics, 2014, 33, 260 CrossRef CAS; (b) S. Sabater, J. A. Mata and E. Peris, Organometallics, 2013, 32, 1112 CrossRef CAS; (c) P. Hauwert, J. J. Dunsford, D. S. Tromp, J. J. Weigand, M. Lutz, K. J. Cavell and C. J. Elsevier, Organometallics, 2013, 32, 131 CrossRef CAS; (d) Y.-C. Lin, H.-H. Hsueh, S. Kanne, L.-K. Chang, F.-C. Liu and I. J. B. Lin, Organometallics, 2013, 32, 3859 CrossRef CAS; (e) A. Chartoire, A. Boreux, A. R. Martin and S. P. Nolan, RSC Adv., 2013, 3, 3840 RSC; (f) Z.-S. Gu, L.-X. Shao and J.-M. Lu, J. Organomet. Chem., 2012, 700, 132 CrossRef CAS PubMed; (g) D. Yuan and H. V. Huynh, Molecules, 2012, 17, 2491 CrossRef CAS PubMed; (h) W. Wang, T. Zhang, F. Wang and M. Shi, Tetrahedron, 2011, 67, 1523 CrossRef CAS PubMed; (i) S. Gonell, M. Poyatos, J. A. Mata and E. Peris, Organometallics, 2011, 30, 5985 CrossRef CAS; (j) L. Gazzola, C. Tubaro, A. Biffis and M. Basato, New J. Chem., 2010, 34, 482 RSC; (k) G. Buscemi, A. Biffis, C. Tubaro, M. Basato, C. Graiff and A. Tiripicchio, Appl. Organomet. Chem., 2010, 24, 285 CAS; (l) G. Buscemi, A. Biffis, C. Tubaro and M. Basato, Catal. Today, 2009, 140, 84 CrossRef CAS PubMed; (m) A. Biffis, L. Gazzola, P. Gobbo, G. Buscemi, C. Tubaro and M. Basato, Eur. J. Org. Chem., 2009, 3189 CrossRef CAS; (n) A. Biffis, C. Tubaro, G. Buscemi and M. Basato, Adv. Synth. Catal., 2008, 350, 189 CrossRef CAS; (o) L. Ray, M. M. Shaikh and P. Ghosh, Organometallics, 2007, 26, 958 CrossRef CAS; (p) M. S. Viciu, E. D. Stevens, J. L. Petersen and S. P. Nolan, Organometallics, 2004, 23, 3752 CrossRef CAS.
- (a) J. J. Dunsford and K. J. Cavell, Organometallics, 2014, 33, 2902 CrossRef CAS; (b) W. Y. Lu, K. J. Cavell, J. S. Wixey and B. Kariuki, Organometallics, 2011, 30, 5649 CrossRef CAS; (c) C. Präsang, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2005, 127, 10182 CrossRef PubMed; (d) W. A. Herrmann, S. K. Schneider, K. Öfele, M. Sakamoto and E. Herdtweck, J. Organomet. Chem., 2004, 689, 2441 CrossRef CAS PubMed; (e) A. Wolfgang, W. A. Herrmann, K. Öfele, S. K. Schneider, E. Herdtweck and S. D. Hoffmann, Angew. Chem., Int. Ed., 2006, 45, 3859 CrossRef PubMed; (f) R. Jazzar, H. Liang, B. Donnadieu and G. Bertrand, J. Organomet. Chem., 2006, 691, 3201 CrossRef CAS PubMed; (g) C. C. Scarborough, B. V. Popp, I. A. Guzei and S. S. Stahl, J. Organomet. Chem., 2005, 690, 6143 CrossRef CAS PubMed; (h) I. Özdemir, S. Demir, B. Cetinkaya and E. Cetinkaya, J. Organomet. Chem., 2005, 690, 5849 CrossRef PubMed.
- (a) E. L. Kolychev, I. A. Portnyagin, V. V. Shuntikov, V. N. Khrustalev and M. S. Nechaev, J. Organomet. Chem., 2009, 694, 2454 CrossRef CAS PubMed; (b) M. Iglesias, D. J. Beetstra, J. C. Knight, L.-L. Ooi, A. Stasch, S. Coles, L. Male, M. B. Hursthouse, K. J. Cavell, A. Dervisi and I. A. Fallis, Organometallics, 2008, 27, 3279 CrossRef CAS; (c) C. C. Scarborough, B. V. Popp, I. A. Guzei and S. S. Stahl, J. Organomet. Chem., 2005, 690, 6143 CrossRef CAS PubMed.
- J. J. Dunsford, K. J. Cavell and B. M. Kariuki, Organometallics, 2012, 31, 4118 CrossRef CAS.
- (a) M. M. Rogers, J. E. Wendlandt, I. A. Guzei and S. S. Stahl, Org. Lett., 2006, 8, 2257 CrossRef CAS PubMed; (b) C. C. Scarborough, M. J. W. Grady, I. A. Guzei, B. A. Gandhi, E. E. Bunel and S. S. Stahl, Angew. Chem., Int. Ed., 2005, 44, 5269 CrossRef CAS PubMed; (c) M. Mayr, K. Wurst, K.-H. Ongania and M. R. Buchmeiser, Chem.–Eur. J., 2004, 10, 1256 CrossRef CAS PubMed.
- C. J. E. Davies, M. J. Page, C. E. Ellul, M. F. Mahon and M. K. Whittlesey, Chem. Commun., 2010, 46, 5151 RSC.
- (a) L. R. Collins, T. M. Rookes, M. F. Mahon, I. M. Riddlestone and M. K. Whittlesey, Organometallics, 2014, 33, 5882 CrossRef CAS; (b) J. K. Park, H. H. Lackey, B. A. Ondrusek and D. T. McQuade, J. Am. Chem. Soc., 2011, 133, 2410 CrossRef CAS PubMed; (c) J. K. Park, H. H. Lackey, M. D. Rexford, K. Kovnir, M. Shatruk and D. T. McQuade, Org. Lett., 2010, 12, 5008 CrossRef CAS PubMed.
- (a) R. Armstrong, C. Ecott, E. Mas-Marzá, M. J. Page, M. F. Mahon and M. K. Whittlesey, Organometallics, 2010, 29, 991 CrossRef CAS; (b) P. S. Kumar, K. Wurst and M. R. Buchmeiser, Organometallics, 2009, 28, 1785 CrossRef CAS; (c) L. Yang, M. Mayr, K. Wurst and M. R. Buchmeiser, Chem.–Eur. J., 2004, 10, 5761 CrossRef CAS PubMed.
- (a) N. Imlinger, K. Wurst and M. R. Buchmeiser, J. Organomet. Chem., 2005, 690, 4433 CrossRef CAS PubMed; (b) P. Bazinet, G. P. A. Yap and D. S. Richeson, J. Am. Chem. Soc., 2003, 125, 13314 CrossRef CAS PubMed.
- Y. Zhang, D. Wang, K. Wurst and M. R. Buchmeiser, J. Organomet. Chem., 2005, 690, 5728 CrossRef CAS PubMed.
- (a) T. Steinke, B. K. Shaw, H. Jong, B. O. Patrick and M. D. Fryzu, Organometallics, 2009, 28, 2830 CrossRef CAS; (b) J. T. Singleton, Tetrahedron, 2003, 59, 1837 CrossRef CAS; (c) S. Gründemann, M. Albrecht, J. A. Loch, J. W. Faller and R. H. Crabtree, Organometallics, 2001, 20, 5485 CrossRef; (d) D. W. Lee, W. C. Kaska and C. M. Jensen, Organometallics, 1998, 17, 1 CrossRef CAS.
- (a) P. L. Arnold, M. S. Sanford and S. M. Pearson, J. Am. Chem. Soc., 2009, 131, 13912 CrossRef CAS PubMed; (b) S. Gu and W. Chen, Organometallics, 2009, 28, 909 CrossRef CAS; (c) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239 CrossRef CAS PubMed; (d) D. Pugh and A. A. Danopoulos, Coord. Chem. Rev., 2007, 251, 610 CrossRef CAS PubMed; (e) J. A. Matá, M. Poyatos and E. Peris, Coord. Chem. Rev., 2007, 251, 841 CrossRef PubMed.
- (a) C.-Y. Liao, K.-T. Chan, J.-Y. Zeng, C.-H. Hu, C.-Y. Tu and H. M. Lee, Organometallics, 2007, 26, 1692 CrossRef CAS; (b) F. E. Hahn, M. C. Jahnke and T. Pape, Organometallics, 2006, 25, 5927 CrossRef CAS; (c) H. M. Lee, J. Y. Zeng, C.-H. Hu and M.-T. Lee, Inorg. Chem., 2004, 43, 6822 CrossRef CAS PubMed; (d) N. Tsoureas, A. A. Danopoulos, A. A. D. Tulloch and M. E. Light, Organometallics, 2003, 22, 4750 CrossRef CAS; (e) W. A. Herrmann, V. P. W. Böhm, C. W. K. Gstöttmayr, M. Grosche, C.-P. Reisinger and T. Weskamp, J. Organomet. Chem., 2001, 617–618, 616 CrossRef CAS; (f) C. Yang, H. M. Lee and S. P. Nolan, Org. Lett., 2001, 3, 1511 CrossRef CAS PubMed.
- A. F. Hill and C. M. A. McQueen, Organometallics, 2012, 31, 8051 CrossRef CAS.
- (a) D. Mercan, E. Çetinkaya and B. Çetinkaya, J. Organomet. Chem., 2011, 696, 1359 CrossRef CAS PubMed; (b) Ì. Özdemir, S. Demir and B. Çetinkaya, ARKIVOC, 2007, xiii, 71 Search PubMed; (c) B. Alici, Ì. Özdemir, N. Gürbüz, E. Çetinkaya and B. Çetinkaya, Heterocycles, 2005, 65, 1439 CrossRef CAS; (d) B. Alici, E. Cetinkaya and B. Cetinkaya, Heterocycles, 1997, 45, 29 CrossRef CAS; (e) E. Cetinkaya, P. B. Hitchcock, A. Hatam and J. Jasim, J. Chem. Soc., Perkin Trans. 1, 1992, 561 RSC; (f) R. F. Ahdulla and R. S. Brinheyer, Tetrahedron, 1979, 35, 1675 CrossRef.
- A. Kumar, M. Katari and P. Ghosh, Polyhedron, 2013, 52, 524 CrossRef CAS PubMed.
- (a) C. Chen, H. Qiu and W. Chen, J. Organomet. Chem., 2012, 696, 4166 CrossRef CAS PubMed; (b) S.-T. Liu, C.-I. Lee, C.-F. Fu, C.-H. Chen, Y.-H. Liu, C. J. Elsevier, S.-M. Peng and J.-T. Chen, Organometallics, 2009, 28, 6957 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of compounds 1–4, DEPT 135 of 4b, COSY of 4b, HSQC of 4b, and characterization data of the products of the catalytic Heck reaction. CCDC 1038971 and 1038972. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra01706h |
| This journal is © The Royal Society of Chemistry 2015 |