
In situ electron beam irradiation-driven formation of quantum dots
Haibo Zeng*abc,
Xiaoming Liab,
Huijie Zhaoab,
Xue Ningab and
Jiayue Xuc
aState Key Laboratory of Mechanics and Control of Mechanical Structures, College of Materials Science and Technology, Nanjing University of Aeronautics and Astronautics, Nanjing 210016, China. E-mail: zeng.haibo@njust.edu.cn
bInstitute of Optoelectronics & Nanomaterials, College of Materials Science and Engineering, Nanjing University of Science and Technology, Nanjing 210094, China
cInstitute of Crystal Growth, School of Materials Science and Engineering, Shanghai Institute of Technology, Shanghai 201418, China
First published on 25th February 2015
Abstract
Recrystallization of amorphous materials is a very interesting phenomenon, but some transformation details are still unknown. Here, we report the formation of ZnO quantum dots (QDs) under electron beam irradiation and a series of in situ consecutive observations on the formation processes. When suffering from the irradiation inside a transmission electron microscope, the amorphous hollow nanoparticles, fabricated through treating the Zn–ZnO core–shell samples, synthesized by laser ablation in liquid with tartaric acid, were found to gradually collapse, and crystallization occurred at the same time. These two isochronous processes collectively induced the formation of the QDs. Interestingly, the size of the QDs can be controlled by the applied precursor particles and beam energy. Numerical simulations demonstrated that the thermal shear stresses greatly stimulate the deformation process, resulting in QD formation.
1. Introduction
Recrystallization of amorphous materials is a very interesting phenomenon, but some transformation details are still unknown. The instability of materials when their dimensions come into the nanoscale becomes a significant issue both for fundamental understanding of nanomaterials and for their applications.1,2 For example, large thermal instability was reported to induce great changes in the electrical conductance of Ga-doped ZnO.3 During an electrocatalysis process, the supported Pt nanoparticle catalysts could also exhibit great instability in terms of their size, shape, and microstructure. This greatly degrades the performance of fuel cells.4 During the chemical vapor deposition growth of Si nanowires, the instability of metal catalyst nanoparticles was reported to largely affect a vapor–liquid–solid process. This could be used to control the product morphology.5 Li et al. pointed out that the instability could be caused by a combined effect of structural disorder and inhomogeneous strain fields.6 Joshi et al. demonstrated that a grain rotation contributes much to the shear instability of nanocrystalline and amorphous materials, and is size-dependent.7 However, it is noted that both theoretical and experimental works which treat the regarded instabilities have been very few to date.On the other hand, amorphous hollow nanoparticles are ideal model materials to investigate the recrystallization of amorphous materials with in situ observation using TEM. Hollow nanostructures have attracted increasing interest because of their specific morphologies and properties that differ from bulky counterparts,8–15 and hence have prominent applications in drug-delivery,16,17 catalysis,18,19 and biologic imaging.20,21 However, due to peculiar structural features, such as hollow interiors and thin shells, they can behave differently with respect to their stability. However, confirmation of such behavior has been lacking till now due to numerous experimental difficulties. Fan et al. observed mass diffusion along the inner pore surfaces in the hollow nanostructures.22,23 Previously, we documented the structural instability of amorphous ZnO hollow nanoparticles (HNPs) during a photocatalysis process. Such instability can be hindered by the intercalation of ultrafine noble metal nanoparticles (NPs) and by improving the ZnO matrix crystallinity.18 On the other hand, recently, in situ processes and synchronous observations using transmission electron microscopy (TEM) have become a very cogent way to investigate the structure and properties of the nanostructures. This technique endows the researcher a “hand” (in situ nanomanipulation) and an “eye” (in situ imaging) at the same time.24–28 Therefore, in situ TEM could provide an effective way to study the crystallization and instability of NPs.
Herein, we present in situ TEM investigations of crystallization and deformation of ZnO HNPs under focused electron beam irradiation. Pronounced instability, manifested by dramatic deformations, was observed during crystallization. This induced a collapse of HNPs and their transformation into monodispersed ZnO quantum dots (QDs) with no specific requirements for a substrate, precise positioning, and facile size control. The features and processes of the deformations were subtly observed, and then the simulated shear stresses within HNPs were thoroughly analyzed. These results would be particularly important to understand small nanostructures grown by a solution method,1,2,18 and could be utilized for diverse nanofabrications.
2. Experiments
2.1. Formation of Zn–ZnO core–shell nanoparticles
The ZnO amorphous HNPs were synthesized by laser ablation in liquid and subsequent weak acid etching. Firstly, a zinc plate (99.99%) was fixed on a bracket in a glass vessel filled with 10 ml 0.05 M sodium dodecyl sulfate (99.5%) aqueous solution. The plate was ablated (irradiated) for 30 min by the first harmonic of a Nd:YAG laser (1064 nm, frequency 10 Hz, pulse duration 10 ns) with a 2 mm focused spot.2.2. Formation of amorphous ZnO hollow nanoparticles
After the formation of nanoparticles by laser ablation in liquid, the 50 ml obtained colloidal solutions were etched by tartaric acid (TA, C4H6O6, 10 mM, 20 ml). The etching agents were slowly dropped into the colloidal solution, droplet by droplet, at the invariable temperature of 40 °C on a stirrer with a heating pedestal. After etching, the colloidal solutions were centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and then ultrasonically re-dispersed in ethanol for more than five cycles to remove the surfactants, and finally draught-dried in an oven at 40 °C for 24 h.
000 rpm and then ultrasonically re-dispersed in ethanol for more than five cycles to remove the surfactants, and finally draught-dried in an oven at 40 °C for 24 h.
2.3. In situ observation on the formation of ZnO quantum dots
The in situ electron beam irradiation experiments were carried out in a high resolution transmission electron microscope (HRTEM, JEOL JEM 2010F) operated at 200 kV. The nanoparticles were re-dispersed in pure ethanol and collected on a TEM copper grid covered with an amorphous carbon film for electron irradiation and HRTEM observations.3. Results and discussion
The starting materials, the ZnO HNPs, were fabricated by laser ablation and subsequent weak acid-etching as discussed in our previous reports.18,29–31 The diameters of the HNPs could be adjusted from 10 to 50 nm under a fabrication process. The small HNPs had an amorphous structure, but the larger HNPs exhibited few crystalline ultrafine grains, which were surrounded by disordered areas within the shells.18,31 The in situ electron beam irradiation was carried out in a HRTEM on HNPs dispersed on carbon-coated grids.3.1. Process of in situ crystallization and deformation
The consecutive morphological evolutions of a single ZnO HNP under electron irradiation were recorded by a series of HRTEM images, as shown in Fig. 1. The primal HNP has a diameter of 12 nm and an intact spherical appearance (Fig. 1(a)). After flash irradiation, a notable structural distortion takes place, especially at the particle surface, Fig. 1(b). Such deformations become more profound with an increasing irradiation time, as seen in Fig. 1(b) and (c). At the same time, mass aggregation occurs in Fig. 1(c), as reflected by the appearance of small dots with a dark contrast. With further irradiation, the mass aggregation becomes more apparent and the discrete new dots become clearly visible, Fig. 1(d). The hollow shell matrix is still preserved. Subsequent irradiation leads to a final collapse of the HNP. The process is accompanied with the appearance of many ultrafine particles, as presented in Fig. 1(e) (30 s irradiation). Additional irradiation promotes particle separation [Fig. 1(f)–(h)]. | ||
| Fig. 1 In situ HRTEM irradiation and imaging of consecutive evolutions of an individual ZnO HNP under irradiation with a focused electron beam. The dashed circles index the area of the starting HNPs. | ||
The changes in crystallinity and composition before and after electron irradiation were then analyzed. The selected area electron diffraction (SAED) pattern of a starting HNP in Fig. 1(a) exhibits faint halos (Fig. 2(a)), indicating an amorphous state, which is in accordance with our previous report.18 After irradiation, (Fig. 1(h)), the SAED pattern taken from the same area exhibits many bright diffraction spots. These form incomplete diffraction rings which can be indexed to wurtzite ZnO in Fig. 2(b). On the other hand, from the EDS spectra in Fig. 2(c), the Zn/O ratios before and after irradiation are 0.91:1 and 0.88:1, respectively, and thus close to the stoichiometric compositions. It is noted that previously, Du et al. reported the crystallization of amorphous SiO2 by ex situ electron beam irradiation and the product was found to be crystalline Si.32 By contrast, Latham et al. reported that Fe oxide compositions were preserved during irradiation-induced crystallization of amorphous Fe oxide NPs.33 Our data show that the Zn oxide reduction does not take place in the present case either.
 | ||
| Fig. 2 Comparison of SAED patterns before (a) and after (b) electron irradiation, and the corresponding EDS spectra (c). | ||
We assume here that the above irradiation-driven evolution may actually contain two underlying processes of in situ crystallization and synchronous HNP deformation leading to monodispersed ZnO QD formation.
The edge area of an HNP was analyzed and is shown in Fig. 3. The disordered state is obvious for the starting HNP, as shown in Fig. 3(a). After short-time irradiation, two grains appeared, denoted as “A” and “B” in Fig. 3(b). During irradiation, they flickered in the field of view and acted as embryo nuclei. At the same time, the contrast of the surrounding area of the grains gradually fades away, which indicates the atomic migration towards the grains. This feeds the grain growth. Under further atomic migration, new grains appear and the former grains grow. On the other hand, such migration towards the grains forms spatial gaps among them, leading to a marked separation, as shown in Fig. 3(c). Moreover, the grain diffusion (described below) should also contribute to the observed morphology changes.
 | ||
| Fig. 3 Local HRTEM observations of the HNP edge area evolutions after 0 s (a), 10 s (b), and 20 s (c) electron irradiations. The dashed circles point out the nascent and grown grains. | ||
3.2. Process features: volume expansion and size effect
From the in situ TEM observations, it is apparent that the crystallization and deformation have two peculiar features, namely, volume expansion and size effect. The former is well reflected by Fig. 1, where the dashed circles point out the areas of the primal HNP. Under electron irradiation, the HNP gradually expands, from Fig. 1(a)–(d). Typically in Fig. 1(d), the primal area becomes notably smaller (after 20 s irradiation) compared to the initial state. The corresponding radius and volume increase to ∼15% and ∼55%, respectively. Considering the preserved integrality of the HNP and such a high expansion ratio, it is believed that the process does not reflect conventional thermal expansion. The collapse of the HNP may thus be regarded as a result of volume expansion under internal stresses. When such stresses exceed a critical value, a ZnO HNP collapses due to its high brittleness and low cohesive energy.On the other hand, the process exhibits significant size effects, as shown in Fig. 4. The sizes of starting the HNPs are 30, 20, and 12 nm (Fig. 4(a)–(c)), whereas the average diameters of the obtained QDs after electron irradiation are 3.4, 2.5, and 1.2 nm (Fig. 4(d)–(f)), respectively. The lattice fringes corresponding to the ZnO (002) planes are visible. The QDs have a narrow size distribution. Noticeably, the sizes of these QDs are comparable with the Bohr diameter of ZnO (1.8 nm). Therefore, the pronounced quantum effects and corresponding changes in optical properties could be envisaged in line with the effective-mass approximation model.34 Compared with the conventional fabrication routes for QDs,35,36 the reported process has several sound advantages. They are namely: (i) no specific requirements for a substrate; (ii) precise QD positioning (the starting HNP could be pre-located), and (iii) a facile size control. These features make the in situ process and the resultant QDs highly useful for optical and optoelectronic nanodevice applications.
 | ||
| Fig. 4 HRTEM images of selected single HNPs with diameter of 30 (a), 20 (b), and 12 (c) nm, and the correspondingly transformed QDs (d)–(f), respectively. | ||
Thermodynamically, the in situ crystallization of the ZnO HNPs was stimulated by the thermal effect of electron beam irradiation, which is similar to amorphous SiO2 (ref. 32) and Fe oxide.33 The irradiation with ∼2 × 106 e s−1 nm−2 intensity could rapidly induce the temperature rise in the materials. The highest temperature for the present ZnO HNPs is estimated to be in the range of 400 to 600 K.37 On the other hand, electron beam heating is reported to decrease the nucleation barrier, which also promotes crystallization.32
The observed HNP deformation under irradiation needs special attention. Although the volume expansion was observed during the amorphous-crystallization of metal glasses due to an increase in “free volume”, the expansion rate documented herein is much larger than those reported for any metal glass.38 The huge volume expansion of the ZnO HNPs could be taken as a macroscopic response to the atomic migrations and grain separations.6 For example, the grain (B) is obviously away from its original position in Fig. 3(c). The outward direction is dominant for the observed volume expansion and deformation. This indicates that the large thermal shear stresses could exist within the HNP shells.
3.3. Simulated shear stress and its size effect
Fig. 5(a) presents the 3-dimensional and planar structural models for the ZnO HNPs, where rin, rout, and r are the inner, outer radii, and the distance to a given mass within the shell, respectively. The applied structural parameters of the HNPs are indexed as (rin, rout). We focused on a site-dependent shear stress, and set the boundary conditions as follows: r = rin, σ = 0 and r = rout, σ = 0, where σ is a thermal stress in the radial direction. According to the thermal elastic theory,39–41 the radial and tangential stresses based on a spherical coordination (dθ = dφ, σθ = σφ) under the equilibrium conditions could be expressed as:
 | (1) |
 | ||
| Fig. 5 Structural models of the HNPs (a), numerically simulated site-dependent thermal shear stresses (b), and diameter-dependent gradients (c). Here, rin, rout, and r are the inner, outer radii, and the distance to a given mass site within the shell. The applied structural parameters of the HNPs are indexed as (rin, rout). | ||
In the case of an external temperature approach, the total thermal strain is written as:
 | (2) |
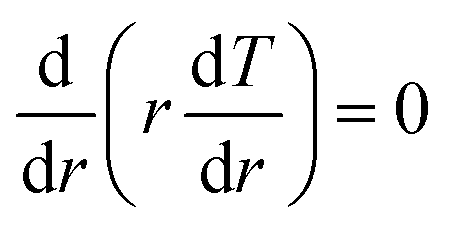 within a spherical shell. Therefore, the stress in the radial direction is deduced as:
within a spherical shell. Therefore, the stress in the radial direction is deduced as:
 | (3) |
Then, a site-dependent thermal shear stress can be simulated as illustrated in Fig. 5(b) according to different structural configurations, and the stress gradients (Fig. 5(c)) can be then obtained from the site-dependent stresses. In the case of the HNPs, the large shear stresses can be induced by electron beam heating. The maximum stresses in the shells are as high as 6–30 MPa, which means that an activation energy up to 0.01 eV should be taken into account. This may excite the observed atomic migrations and the diffusion of the grains under crystallization and deformation processes. More importantly, the stress gradients can be as high as 6–28 MPa, as shown in Fig. 5(c), which could induce inhomogeneity of the response of the structure to electron beam heating. This supports the morphological evolution of the HNPs, as observed in Fig. 1 and 3. Similarly, Spaepen demonstrated that small thermal fluctuations could break any existing defect in an amorphous metal into small segments with lower configuration energies.42
For the HNPs with different structural configurations, shear stresses exhibit obvious size effects. Through comparing the curves in Fig. 5(b), one can see that the shear stress intensity decreases with an increase in rin and rout. For example, the stress decreases from 29.4 to 8.8 MPa with an increase in the radius from 6 to 14 nm. The stress gradients also decrease from 27.4 to 5.6 MPa nm−1 with enlargement of the original HNP size. The decreased intensity and gradient of the shear stress would depress the activity of atomic migration and grain diffusion. Such changes in the larger HNPs would make the deformation process milder during crystallization, and advance the grains to grow larger in the thermodynamic conditions with higher equilibrium. This is in accordance with a size decrease of the resulting QDs from the smaller starting HNPs, as observed in Fig. 4. Previously, Joshi and Ramesh theoretically treated the size effects of a grain rotation, which induced the shear instability in the nanocrystalline and amorphous materials.7
Furthermore, it is worth noting that the size effects of shear stresses and their gradients are much more significant for the small size range. With decreasing size, the increasing rates of the intensity and the gradient of the shear stress become abrupt (i.e. when the radius is below 20 nm in Fig. 5(b) and (c)). By contrast, these rates are nearly saturated and only marginally changeable in the large size range. These phenomena demonstrate that the instability will become more dominant in smaller HNPs, where a stronger deformation should occur during crystallization. Li’s simulations also showed that a disorder and internal inhomogeneous strain fields play an important role in the instability.6 Recently, we have also observed the significant enhancement of instability of ZnO and ZnS nanowires when the concentration of dopants was in a certain range, which also induces the local disorder and stress–strain fields within a lattice.43
4. Conclusions
In summary, in situ HRTEM observations have been performed to explore the crystallization and deformation of ZnO HNPs under focused electron beam irradiation. A series of significant morphological changes were observed. These resulted in the collapse of the HNPs and their transformation into monodispersed ZnO QDs. Compared with conventional routes, the present QD formation process has several sound advantages such as no specific requirements for a substrate, precise QD positioning and size control. The detailed observations demonstrate that the atomic migration and separation of the grains are the kinematic inducements of the dramatic HNP deformations under crystallization, accompanied by huge HNP volume expansion and notable size effects. The numerical simulations demonstrate that the thermal shear stresses and their gradients greatly stimulate the atomic migrations and diffusion of the grains, and hence the dramatic deformations. The size effects of the calculated thermal shear stresses presume their dominance for the small-sized HNPs and for those exhibiting an amorphous or partially disordered state. These would induce larger deformations during a crystallization process and could be utilized for smart QD nanofabrication.Acknowledgements
This work was supported by the National 973 project from National Basic Research Program of China (2014CB931700/2014CB931702), the National Natural Science Foundation of China (61222403), the Program for Eastern Scholar at Shanghai Institutions of Higher Learning (2012-53), and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.References
- M. Li and W. L. Johnson, Phys. Rev. Lett., 1993, 70, 1120 CrossRef PubMed.
- X. M. Bai and M. Li, Nano Lett., 2006, 6, 2284 CrossRef CAS PubMed.
- J. A. Sans, G. M. Criado, J. P. Porres, J. F. S. Royo and A. Segura, Appl. Phys. Lett., 2007, 91, 221904 CrossRef.
- Y. S. Shao-Horn, C. Sheng, W. S. Chen, P. J. Ferreira and D. Morgan, Top. Catal., 2007, 46, 285 CrossRef CAS.
- L. Cao, B. Garipcan, J. S. Atchison, C. Ni and J. E. Spanier, Nano Lett., 2006, 6, 1852 CrossRef CAS PubMed.
- G. P. Zheng and M. Li, Acta Mater., 2007, 55, 5464 CrossRef CAS.
- S. P. Joshi and K. T. Ramesh, Phys. Rev. Lett., 2008, 101, 025501 CrossRef PubMed.
- Y. D. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes and A. P. Alivisatos, Science, 2004, 304, 711 CrossRef CAS PubMed.
- P. X. Gao and Z. L. Wang, J. Am. Chem. Soc., 2003, 125, 11299 CrossRef CAS PubMed.
- M. S. Mo, J. C. Yu, L. Z. Zhang and S. K. A. Li, Adv. Mater., 2005, 17, 756 CrossRef CAS.
- M. S. Mo, S. H. Lim, Y. W. Mai, R. K. Zheng and S. P. Ringer, Adv. Mater., 2008, 20, 339 CrossRef CAS.
- F. Q. Sun and J. C. Yu, Angew. Chem., Int. Ed., 2007, 46, 773 CrossRef CAS PubMed.
- G. Z. Shen, Y. Bando, C. H. Ye, X. L. Yuan, T. Sekiguchi and D. Golberg, Angew. Chem., Int. Ed., 2006, 45, 7568 CrossRef CAS PubMed.
- K. An, S. G. Kwon, M. Park, H. B. Na, S. Baik, J. H. Yu, D. Kim, J. S. Son, Y. W. Kim, H. M. Park and T. Hyeon, Nano Lett., 2008, 8, 4252 CrossRef CAS PubMed.
- H. J. Fan, U. Gösele and M. Zacharias, Small, 2007, 3, 1660 CrossRef CAS PubMed.
- E. Mathiowitz, J. S. Jacob, Y. S. Jong, C. A. Santos, K. Vijayaraghavan, S. Montgomery, M. Bassett and C. Morrell, Nature, 1997, 386, 410 CrossRef CAS PubMed.
- T. Yamada, Y. Iwasaki, H. Tada, H. Iwabuki, M. K. L. Chuah, T. VandenDriessche, H. Fukuda, A. Kondo, M. Ueda and M. Seno, Nat. Biotechnol., 2003, 21(8), 885 CrossRef CAS PubMed.
- H. B. Zeng, W. P. Cai, P. S. Liu, X. X. Xu, H. J. Zhou, C. Klingshirn and H. Kalt, ACS Nano, 2008, 2, 1661 CrossRef CAS PubMed.
- B. X. Li, Y. Xie, M. Jing, G. X. Jing, G. X. Rong, Y. C. Tang and G. Z. Zhang, Langmuir, 2006, 22, 9380 CrossRef CAS PubMed.
- C. H. Su, H. S. Sheu, C. Y. Lin, C. C. Huang, Y. W. Lo, Y. C. Pu, J. C. Weng, D. B. Shieh, J. H. Chen and C. S. Yeh, J. Am. Chem. Soc., 2007, 129, 2139 CrossRef CAS PubMed.
- J. Chen, F. Saeki, B. J. Wiley, H. Cang, M. J. Cobb, Z. Y. Li, L. Au, H. Zhang, M. B. Kimmey, X. Nan and D. Li, Nano Lett., 2005, 5, 473 CrossRef CAS PubMed.
- H. J. Fan, M. Knez, R. Scholz, D. Hesse, M. Zacharias and U. Gösele, Nat. Mater., 2006, 5, 627 CrossRef CAS PubMed.
- H. J. Fan, M. Knez, R. Scholz, D. Hesse, K. Nielsch, M. Zacharias and U. Gösele, Nano Lett., 2007, 7, 993 CrossRef CAS PubMed.
- D. Golberg, P. M. F. J. Costa, M. Mitome and Y. Bando, J. Mater. Chem., 2009, 19, 909 RSC.
- Y. Gao and Y. Bando, Nature, 2002, 415, 599 CrossRef CAS PubMed.
- H. G. Duan, E. Q. Xie, L. Han and Z. Xu, Adv. Mater., 2008, 20, 3284 CrossRef CAS.
- Y. J. Gan and F. Banhart, Adv. Mater., 2008, 20, 4751 CrossRef CAS.
- Y. Yang, R. Scholz, A. Berger, D. S. Kim, M. Knez, D. Hesse, U. Gösele and M. Zacharias, Small, 2008, 4, 2112 CrossRef CAS PubMed.
- H. B. Zeng, W. P. Cai, Y. Li, J. L. Hu and P. S. Liu, J. Phys. Chem. B, 2005, 109, 18260 CrossRef CAS PubMed.
- H. B. Zeng, Z. G. Li, W. P. Cai, B. Q. Cao, P. S. Liu and S. K. Yang, J. Phys. Chem. B, 2007, 111, 14311 CrossRef CAS PubMed.
- H. B. Zeng, P. S. Liu, W. P. Cai, S. K. Yang and X. X. Xu, J. Phys. Chem. C, 2008, 112, 19620 CAS.
- X. W. Du, M. Takeguchi, M. Tanaka and K. Furuya, Appl. Phys. Lett., 2003, 82, 1108 CrossRef CAS.
- A. H. Latham, M. J. Wilson, P. Schiffer and M. E. Williams, J. Am. Chem. Soc., 2006, 128, 12632 CrossRef CAS PubMed.
- Y. Kayanuma, Phys. Rev. B, 1988, 38, 9797 CrossRef.
- Y. S. Fu, X. W. Du, S. A. Kulinich, J. S. Qiu, W. J. Qin, R. Li and J. Liu, J. Am. Chem. Soc., 2007, 129, 16029 CrossRef CAS PubMed.
- J. G. Lu, Z. Z. Ye, Y. Z. Zhang, Q. L. Liang, S. z. Fujita and Z. L. Wang, Appl. Phys. Lett., 2006, 89, 023122 CrossRef.
- M. Liu, L. Y. Xu and X. Z. Lin, Scanning, 1994, 16, 1 CrossRef CAS.
- Q. K. Li and M. Li, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 094101 CrossRef.
- G. Ouyang, X. L. Li and G. W. Yang, Appl. Phys. Lett., 2007, 91, 051901 CrossRef.
- G. Ouyang, X. L. Li, X. Tan and G. W. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 193406 CrossRef.
- C. Q. Sun, Prog. Solid State Chem., 2007, 35, 1 CrossRef CAS.
- F. Spaepen, Defects in Amorphous Solids, Les Houches Lectures on Physics of Defects, North-Holland, Amsterdam, 1981, pp. 133–174 Search PubMed.
- B. Liu, Y. Bando, X. Jiang, C. Li, X. Fang, H. B. Zeng, T. Terao, C. Tang, M. Mitome and D. Golberg, Nanotechnology, 2010, 21, 375601 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |