
Compatibilization strategies in poly(lactic acid)-based blends
Jian-Bing Zeng
*a,
Kun-Ang Li
a and
An-Ke Du
b
aCollege of Chemistry and Chemical Engineering, Southwest University, Beibei, Chongqing 400715, China. E-mail: jbzeng@swu.edu.cn; Fax: +86-23-68254000; Tel: +86-23-68254000
bChongqing Academy of Science and Technology, Chongqing 401123, China
First published on 23rd March 2015
Abstract
Poly(lactic acid) (PLA) is regarded as one of the most promising bio-based and biodegradable polymers, due to its excellent biodegradability, biocompatibility, renewability, high strength, and easy processibility. However, disadvantages such as brittleness and relatively high cost have restricted its applications significantly. Polymer blending provides an economic and efficient way to modify the properties of PLA. Most shortcomings of PLA are theoretically surmountable by blending with abundant polymers with various properties. But, unfortunately, PLA is thermodynamically immiscible with most existing polymers. High-performance PLA-based blends are usually unanticipated by direct blending. In order to obtain PLA-based blends with excellent overall properties, compatibilization is required during polymer blending. Various strategies have been employed or developed to compatibilize PLA blends with different polymers, as reported in recent studies. This article aims to review the development in compatibilization strategies employed in PLA-based blends.
 Jian-Bing Zeng | Jian-Bing Zeng earned his PhD in the College of Chemistry at Sichuan University in 2009. He served as an associate professor at Sichuan University in 2011. He has been a professor in the College of Chemistry and Chemical Engineering at Southwest University (Chongqing, China) since 2014. His research interests are biopolymers and their nanocomposites. |
 Kun-Ang Li | Kun-Ang Li will finish his undergraduate study for a material chemistry degree from the College of Chemistry and Chemical Engineering, Southwest University in 2015. He has been in charge of scientific research projects several times during his college career from school to state level. His research interests concern biodegradable polymer nanocomposites. |
 An-Ke Du | An-Ke Du earned his PhD in the College of Chemistry at Sichuan University in 2011 and worked as a postdoctoral researcher at Chongqing University in 2012. He has been an assistant professor in the Chongqing Academy of Science and Technology since 2012. His research interests focus on polymer modification. |
1. Introduction
Biodegradable polymers especially those derived from renewable resources have attracted great attention due to the increasing environmental concerns and resource crisis associated with traditional petroleum-based polymers.1–3 Poly(lactic acid) or polylactide (PLA) is one of the most extensively investigated biobased and biodegradable polymers due to its attractive mechanical strength, high melting temperature, excellent biodegradability, sustainability, and relatively low cost.4 The monomer for PLA is lactic acid, which was first isolated from sour milk by Scheele in 1780 and first commercially available in 1881.5 The majority of the world's commercial lactic acid is now made by bacterial fermentation of saccharides, and various technologies for purification of lactic acid were reported in a review by Datta and Henry.6 In 1845, Pelouze for the first time synthesized PLA by condensation of lactic acid under continuous removal of water.7 However, applicable PLA with high molecular weight is difficult to be produced by this method due to the low reactivity of lactic acid and the reversibility of the polymerization technique.Commercial PLA is often produced by ring-opening polymerization (ROP) of lactide, the dimer of lactic acid, prepared by depolymerization of low molecular weight PLA oligomer,4 as shown in Fig. 1. The purity of lactide is very important for synthesizing high molecular weight PLA. Carothers and coworkers for the first time prepared PLA by ROP of lactide,7 but high molecular weight PLA was only obtained after DuPont developed purification techniques in 1954.4 Recently, Cargill Dow LLC has commercialized PLA under the trade name NatureWorks at a capacity of 140![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons per year with starch as the starting material in 2002.8 Fermentation of starch gives rise to lactic acid, condensation of lactic acid leads to PLA oligomer, and the catalytic depolymerization of the oligomer under vacuum produces lactide. After purification, high molecular weight PLA could then be synthesized by ROP of lactide in the presence of a catalyst such as Sn(Oct)2.4 Besides ROP, some other techniques such as chain-extension reaction,9–11 azeotropic dehydration condensation,12,13 and melt/solid state polymerization14,15 could also be used to prepare high molecular weight PLA with values of more than 100000 g mol−1. The various synthetic routes for the preparation of high molecular weight PLA are schematically shown in Fig. 1.
000 tons per year with starch as the starting material in 2002.8 Fermentation of starch gives rise to lactic acid, condensation of lactic acid leads to PLA oligomer, and the catalytic depolymerization of the oligomer under vacuum produces lactide. After purification, high molecular weight PLA could then be synthesized by ROP of lactide in the presence of a catalyst such as Sn(Oct)2.4 Besides ROP, some other techniques such as chain-extension reaction,9–11 azeotropic dehydration condensation,12,13 and melt/solid state polymerization14,15 could also be used to prepare high molecular weight PLA with values of more than 100000 g mol−1. The various synthetic routes for the preparation of high molecular weight PLA are schematically shown in Fig. 1.
 | ||
| Fig. 1 Synthetic routes to poly(lactic acid). | ||
Due to the presence of two chiral carbon centers, lactide has three stereoisomers: D,D-lactide (D-LA), L,L-lactide (L-LA), and D,L-lactide (meso-LA), as shown in Fig. 2. The physical properties including melting temperature, crystallization behaviors, and mechanical properties of PLA depend strongly on the stereochemical compositions. PLA homopolymer polymerized from pure L-LA or D-LA has an equilibrium crystalline melting point of 207 °C.8 However, commercially available PLA usually shows a melting point of 170–180 °C due to the slight racemization, imperfect crystallites, and impurities.4 A 1:1 mixture of poly(L-lactide) and poly(D-lactide) presents a higher melting temperature of 230 °C and better mechanical properties than either homopolymer due to the formation of a stereocomplex structure.16,17 The effect of stereochemical composition on the glass transition temperature is much less significant than on the melting temperature as crystalline PLA and amorphous PLA show similar glass transition temperature of 55–63 °C.4 Although there are three types of PLA, commercial PLA is a copolymer of poly(L-lactide) with small amount of poly(D,L-lactide), since the lactic acid derived from biological sources is composed of mostly L-lactic acid and smaller amount of D-lactic acid.13 In the following sections, for brevity, the stereo structure of PLA is not discriminated since the compatibilization techniques in PLA blends are almost the same regardless of the stereo structure.
 | ||
| Fig. 2 Chemical structures of three stereoisomers of lactide. | ||
PLA shows many advantages which make it widely used in many fields. Besides being derived from renewable resources (e.g., corn, sugar, potato), PLA is recyclable,18,19 biodegradable and compostable with the final degradation products of carbon dioxide,20–22 and easily processible to form applicable products with traditional processing equipment.13,23 These characteristics make PLA very attractive to replace non-degradable petroleum-based plastics in commodity plastic applications, such as mulch film, disposable cutlery, shopping bags, trash bags, food containers, and packaging materials.13,23–25 The good biocompatibility and bioresorbability enable PLA to find extensive applications in biomedical and pharmaceutical fields including surgical sutures, tissue engineering materials, and drug delivery systems.26 The high strength and melting temperature help PLA to find potential application in engineering plastics.27 The tensile strength of PLA is usually in the range of 50–70 MPa depending on the molecular weight and stereochemical composition, and the Young's modulus can be as high as 3 GPa.28
However, there are still some drawbacks that restrict the wide application of PLA. For example, PLA is lacking in toughness and has very low impact strength and short extensibility, which are some of the biggest problems that restrict the use of PLA in many areas, where good impact resistance is required. The elongation at break is usually less than 10% and the impact strength is only ∼2.5 kJ m−2.28,29 In addition, the poor crystallizability, slow biodegradation rate, and low heat distortion temperature are the other shortcomings limiting the wide application of PLA.8 In order to extend the application of PLA, modifications have to be done to improve the properties.
2. PLA modifications
The most widely used methods to modify the properties of polymers include chemical copolymerization, polymer blending, and nanocomposite technology. Chemical copolymerization is a very important way of modifying properties of homopolymers, and a variety of commercially important copolymers have been achieved via macromolecular design and chemical copolymerization. With respect to structure–properties relationships, new materials with tunable properties can be prepared by judicious selection of co-monomers and the variation of copolymer compositions. Physical blending is a convenient route for developing new polymeric materials, which combine the advantages of more than one existing polymer. The properties of the resulting polymer blends are also tunable through the choice of blending partners and the change of blend compositions. Nanocomposite technology involves the nanoscale dispersion of nanosized fillers into a polymer matrix. The nanofillers have very high surface areas. With good dispersion, their high surface area could potentially lead to reinforced properties of the nanocomposites. The reinforcing efficiency is usually better than that of conventional micro- and macro-composites for the same quantity of fillers. We do not introduce this technique of property modification of PLA in any detail, although this method provides an efficient way of reinforcing physical properties without sacrificing the advantages of the polymer matrix. Detailed information for property modification of PLA through nanocomposite technology can be found in a recent review by Raquez et al.30 However, the use of nanoparticles as compatibilizers for PLA-based blends is included in the review. In the following text, we briefly introduce the modification of PLA via chemical copolymerization and physical blending.2.1. Chemical copolymerization
For property modification or developing new materials, PLA has been copolymerized with a variety of polymers including polyesters, polyolefins, and natural polymers through several polymerization techniques such as condensation polymerization,31,32 ROP,33–35 and chain-extension reaction.10,11,36 Herein, we do not review this technique in detail, but briefly summarize its advantages and disadvantages. Detailed information for property modification of PLA via chemical copolymerization can be found in a recent review paper by Rasal et al.37 The greatest advantage of chemical copolymerization should be that there are abundant materials with various properties that can be selected to copolymerize with PLA to generate a variety of new materials with tunable properties thus having versatile applications. Materials from bio-based to petroleum-based, biodegradable to non-degradable, crystalline to amorphous can be used to copolymerize with PLA to produce novel materials with various properties. However, the disadvantages of chemical copolymerization for modification of PLA are also conspicuous. The improvement in certain properties of one polymer achieved by chemical copolymerization is always accompanied by the deterioration of other properties. Taking the toughening of PLA by copolymerization with poly(ε-caprolactone) (PCL) as an example, the elongation at break of PLA–PCL multiblock copolymer could reach 600%, depending on the composition; however, the tensile strength and Young's modulus drop dramatically to 32 MPa and 30 MPa, respectively, and the melting temperature and degree of crystallinity are also apparently decreased.38 The lengthy reaction period, rigorous copolymerization conditions and high cost constitute the other disadvantages with respect to modification of PLA via chemical copolymerization especially through ROP. In a word, chemical copolymerization is a powerful technique for developing new materials with novel properties and applications but not a convenient and economic method of modifying the properties of PLA without significant loss of other properties.2.2. Polymer blending
In contrast to chemical copolymerization, physical blending with a carefully selected component represents an economic and convenient way of modifying properties of homopolymers.39–41 To develop new materials with desired properties, PLA has been blended with various plasticizers and polymers.8,28,29 The introduction of small molecular or macromolecular plasticizers would significantly improve the toughness especially the elongation at break of PLA due to the plasticization effect, which could reduce the glass transition temperature and thus increase the ductility of PLA.28,29 More importantly, physical blending with other polymers provides the most promising way to modify the properties of PLA. PLA-based materials with a wide range of properties are theoretically obtainable by blending since a great number of polymers with various properties can be selected to blend with PLA. However, this does not mean that excellent properties of PLA blends are easily obtainable by simple blending. On the contrary, it is difficult to prepare high-performance PLA blends through simple blending in most cases since most of the existing polymers are incompatible with PLA.42–48 Compatibilization is usually required for the incompatible polymer blends to exhibit excellent properties.3. Compatibilization strategies
3.1. Generalities of compatibilization
Before going on to describe compatibilization, it is necessary to talk about miscibility. Miscibility is a thermodynamic term that describes the behavior of a polymer pair by specifying the number of phases and their composition that are formed upon blending.41 There are three types of blends in terms of miscibility: (1) completely miscible, (2) partially miscible, and (3) fully immiscible. The miscibility of a polymer blend can be distinguished from the morphology and the glass transition temperature (Tg) of the blend. Two types of morphologies, i.e., homogeneous and heterogeneous, exist for polymer blends. A completely miscible blend exhibits a homogeneous morphology with a single Tg, which is between the Tg values of both components and changes with the composition. A partially miscible blend, usually presenting a fine phase morphology with improved properties, is referred to as a compatible blend.41 Two phases exist in a partially miscible blend and each phase is homogeneous with a part of one polymer dissolved in the other. This blend has two Tg values corresponding to the two phases, and each Tg shifts from the value of one component towards that of the other. A fully immiscible blend usually exhibits a macrophase-separated morphology with coarse interface boundary, large dispersed phase size, poor phase adhesion, and two Tg values, which are independent of blend compositions.The state of miscibility of two polymers is governed by the free energy of mixing, ΔGmix, which is defined as
| ΔGmix = ΔHmix − TΔSmix |
Compatibility is a technical term defining the phase morphology and property profile of a blend in view of a certain application.50 If blending of two partially miscible or immiscible polymers generates a fine phase morphology and combines advantageous properties of the blend components, the compatibility between the two polymers is good, while they are incompatible if blending results in coarse phase morphology and poor properties. For the incompatible polymer blends, their compatibility can be improved via an appropriate method, which is usually referred to as compatibilization. We say that the compatibility of an incompatible blend is changed to compatible if the phase morphology transfers from coarse to fine and the properties change from poor to good after compatibilization.
Compatibilization is a technique to improve compatibility and enhance properties of immiscible polymer blends. The most important roles of compatibilization are first to reduce the size of the dispersed phase through the reduction of interfacial tension and second to prevent the dispersed phase from coalescence, thus to stabilize the formed fine phase morphology.41 In addition, compatibilization can improve the interfacial interactions between dispersed phase and matrix as a result of using compatibilizers that are usually macromolecular species showing interfacial activities in heterogeneous blends.50 With formation of fine phase morphology and improved interfacial interaction, useless incompatible blends can be changed to useful compatible materials which combine the excellent properties of the blend components. Taking immiscible poly(lactic acid)/low-density polyethylene (PLLA/LDPE) blend as an example, Fig. 3 shows the morphologies of cryofractured surfaces for immiscible PLLA/LDPE before and after being compatibilized by block copolymer PLLA-b-LDPE, as reported by Wang and Hillmyer.42 The uncompatibilized blend showed coarse morphology with large LDPE dispersed particles and obvious phase boundary, indicating poor compatibility and interfacial adhesion, while the size of dispersed LDPE particles decreased gradually and the phase boundary became less distinct with an increase in the content of compatibilizer. The compatibilized blends showed significantly improved mechanical properties over those of the pristine blend. In addition to the use of block copolymers, there are several other approaches that can compatibilize PLA-based blends, as described in the following section.
 | ||
| Fig. 3 Morphologies for cryofractured surfaces of (a) 80:20 PLLA/LDPE, and (b) 80:20:2, (c) 80:20:5, and (d) 80:20:10 PLLA/LDPE/PE-b-PLLA blends. Reprinted with permission from ref. 42. Copyright 2001 John Wiley. | ||
3.2. Addition of premade copolymers
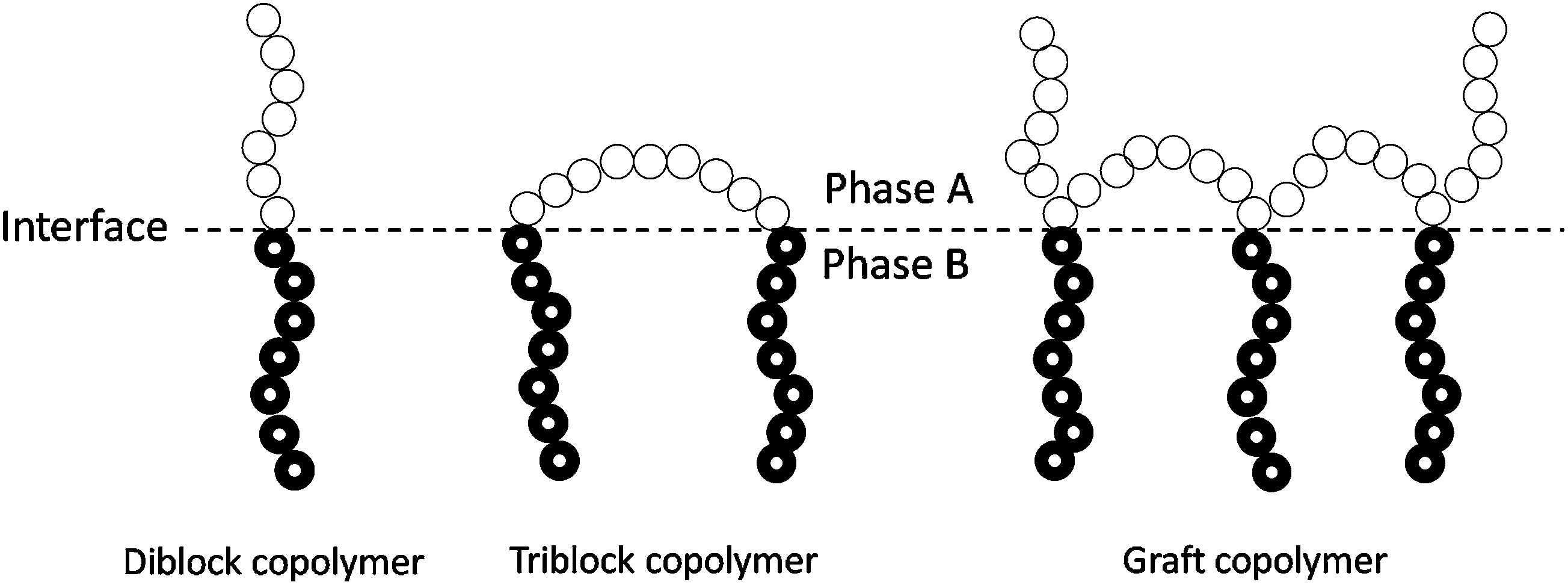 | ||
| Fig. 4 Ideal location of diblock, triblock, and graft copolymers at the interface of an immiscible A/B polymer blend. | ||
The presence of the block or grafted copolymers at the interface can decrease the interfacial tension of the immiscible blends, thus reducing the size of the droplets of dispersed phase during melt processing.41 Usually, the minor phase exhibits an average particle size in the sub-micrometer range when dispersed in the other polymer matrix. In addition, the existence of the blocky-structured copolymer at the surface could prevent coalescence of the generated dispersed particles during subsequent processing or storage. Therefore, the addition of blocky-structured copolymers as compatibilizers is able to form and stabilize a fine phase morphology in phase-separated polymer blends. It is worth noting that the presence of the blocky-structured copolymers can enhance the interfacial adhesion of the immiscible blends due to the entanglement of each block with the corresponding blend component. Sufficient interfacial adhesion is essential for stress transfer from one phase to the other, which is efficient in stopping the cracks initiated at the interface from growth to catastrophic failure. The formation and stabilization of a fine phase morphology and the improvement in the interfacial adhesion usually change a useless immiscible blend to a useful material in which the advantages of each blend component are combined.41
Bai et al.54 reported the use of poly(D,L-lactide-co-p-dioxanone) (PLADO) random copolymer to compatibilize a poly(p-dioxanone)/poly(D,L-lactide) (PPDO/PLA, 80/20 w/w) blend, and found that the addition of PLADO could obscure the phase boundary between PPDO and PLA phases and increase the compatibility between the two components, although the average number of sequential comonomeric units of PDO unit was only 1.0. Recently, they used poly(p-dioxanone-co-L-lactide) (PDOLLA) to compatibilize a PLLA/PPDO (85/15 w/w) blend, and found that the compatibility and mechanical properties of the blend were much improved with addition of 3.0 wt% PDOLLA.55 In order to improve the compatibility of a poly(D,L-lactic acid)/poly(glycolic acid) (PLA/PGA) blend, Ma et al.56 added poly(D,L-lactic acid-co-glycolic acid) (PLAGA) as a compatibilizer to the mixed solution of PLA and PGA during film preparation, and found that when 5% PLAGA was added the blend showed a smooth and homogeneous morphology which was similar to that of neat PGA film.
PLA–PCL diblock or triblock copolymers have been widely used to compatibilize immiscible PLA/PCL blends. Choi et al.53 synthesized a PLA–PCL diblock copolymer and used it to compatibilize PLA/PCL blends and found that the size of PCL domains in PLA matrix can be reduced upon addition of PLA–PCL diblock copolymer; however, the extent of reduction was less than caused by the addition of PLA–PCL random copolymer. Maglio et al.58–60 and Wu et al.61 used a PLA–PCL–PLA triblock copolymer to compatibilize PLA/PCL blends. The good emulsifying effect was evidenced by the strong reduction in particle size of dispersed PCL phase upon addition of the triblock copolymer. For example, the dimension of dispersed PCL domains in PLA/PCL (70/30, w/w) drastically decreases from about 10–15 μm to about 3–4 μm after adding 4 wt% of the triblock copolymer.59
To improve the compatibility of PLA with natural rubber, Chumeka et al.62 synthesized a diblock copolymer from hydroxyl telechelic natural rubber (NR) oligomers and PLA and used it as a compatibilizer for PLA/NR blend. The results showed that the size of dispersed particles was reduced by the addition of the diblock copolymer.
Na et al.48 have extended this approach to C–B block copolymers, where the C block was miscible with PLA. They blended PLA with PEG-b-PCL block copolymer and found that PLA was miscible with the PEG block while immiscible with the PCL block although it was block-copolymerized with PEG. Then they employed this block copolymer to compatibilize PLA/PCL blends and achieved improved mechanical properties upon addition of the copolymer. Considering that polyoxyethylene (PEO) is miscible with both PLA and PCL, the diblock copolymer PLA-b-PEO of “A–C” type was employed by Maglio et al. to compatibilize PLA/PCL blend and it exhibited similar behavior to PLA-b-PCL-b-PLA triblock copolymer.60 Chang et al.63 improved compatibility between PLLA and soybean oil (SOY) by addition of poly(isoprene-b-lactide) (A–C type) in which polyisoprene is miscible with SOY due to the small Flory–Huggins interaction parameter. With the aid of poly(isoprene-b-lactide), the incorporated content of SOY could be increased from 6 wt% to 20 wt%, and a phase inversion occurred with the minor SOY changing to matrix surrounding PLLA particles to provide improved toughness.
Another type of copolymer (C–D type) such as polyethylene oxide–polypropylene oxide–polyethylene oxide (PEO–PPO–PEO) triblock copolymer is sometimes used to compatibilize PLLA blends. For example, PEO–PPO–PEO was employed as a compatibilizer in PLLA/PCL64 and PLLA/PBSL65 blends. In those systems, PEO–PPO–PEO worked like a third homopolymer such as PEG which was miscible with both blend components and thus could emulsify the phase interface to improve compatibility. Real C–D type block copolymers, of which one block (C block) is miscible with A component and the other block (D block) is miscible with B component, have seldom been used in compatibilization of PLLA blends, possibly due to the difficulty of finding such specific block copolymers.
From the above description, we can see that addition of premade copolymers is an efficient way to compatibilize PLA blends with various polymers. Besides the powerful compatibilization efficiency, the best advantage of this technique is its universality. Theoretically, this method can be used to compatibilize various immiscible blends by careful design and synthesis of suitable copolymers. The literature that focuses on the use of this method to compatibilize immiscible blends grows rapidly. However, this technique seems not to be suitable in large-scale production due to the commercial unavailability and high cost of the specific copolymers.
3.3. Addition of reactive polymers
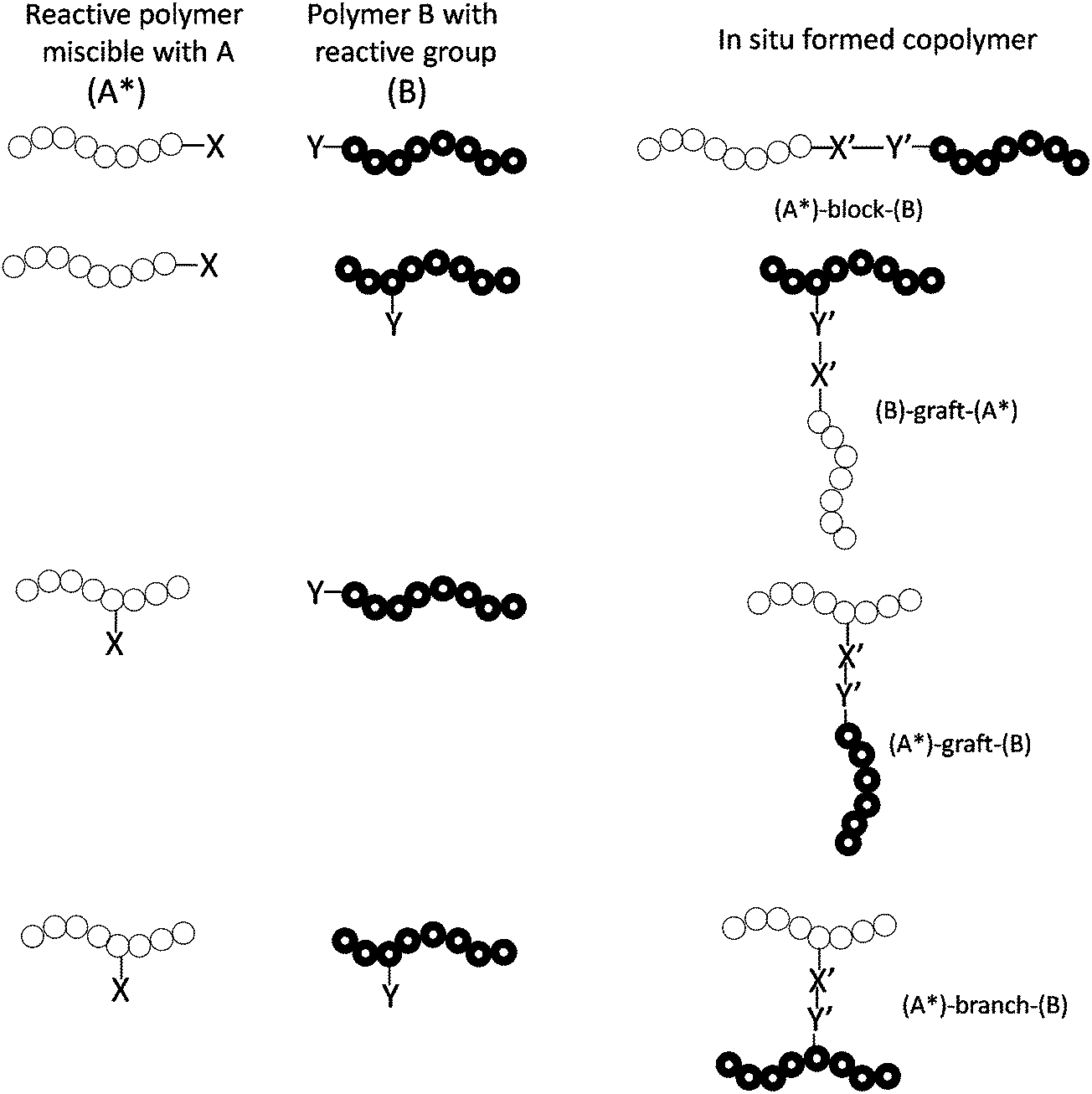 | ||
| Fig. 5 Reactive polymer and the type of copolymer formed during processing. | ||
There are many advantages of addition of reactive polymers over addition of premade copolymers. Firstly, the reactive polymers only give rise to block or graft copolymers at the location where they are needed, always at the interface of immiscible blends, which should be more efficient in compatibilization than the addition of premade copolymers. Secondly, reactive polymers usually show lower melt viscosity than premade copolymers, at least if the blocks of premade copolymer possess similar molecular weight to the reactive “blocks”, which makes the reactive polymer diffuse towards the interface of immiscible blends much faster than the premade copolymer. This is extremely important with respect to the short processing time during reactive blending which is usually of the order of a minute or even less. In some cases, the reactive polymers may not be miscible with either component of the blend, but they can also be used as compatibilizers if they are reactive towards the functional groups of both blend components. Copolymers, working as compatibilizers, could also be formed at the interface of immiscible blends through the reaction between the components in the presence of the reactive polymers.68
In order to successfully compatibilize immiscible blends with reactive polymers, they must have a suitable reactivity with the functional groups of the blend components so as to accomplish reaction to form block or graft copolymers during the short blending time. Furthermore, the formed covalent bonds must be stable enough to remain intact under the subsequent processing conditions. PLA is an aliphatic polyester with terminal groups of carboxyl and hydroxyl, which are reactive with many functional groups such as epoxy, anhydride, isocyanate, and oxazoline groups. Fig. 6 shows the reactions between terminal groups of PLA and reactive polymers with those functional groups.
 | ||
| Fig. 6 Reactions between functional groups of reactive polymers and terminal groups of PLA. | ||
Addition of reactive polymers to compatibilize immiscible blends also has some other advantages. Compared to graft and block copolymers, reactive polymers with various functional groups are easier to produce with simple techniques and some reactive polymers even have been commercialized. However, this technique also has some disadvantages. The reactive polymers with functional groups may be poisonous, which would cause some potential injuries to operators. The inevitable residue of some poisonous functional groups if remaining would cause some safety problems to the resulting blends. Nevertheless, this method has been widely used in PLA-based blends. Many reactive polymers with various functional groups that were used in compatibilization of PLA-based blends are described in the following text.
Poly(butylene adipate-co-terephthalate) (PBAT) is a flexible biodegradable polyester that can be used to toughen PLA without compromising the biodegradability. Zhang et al.68 improved the compatibility between PLA and PBAT by adding a polymer containing 8% GMA, which, containing epoxy groups, can react with both PLA and PBAT to form copolymers of PLA and PBAT and thus to compatibilize the blends. Al-Itry et al.72 also compatibilized PLA/PBAT blends with a reactive polymer named Joncryl containing nine GMA functions.
Natural rubber (NR) is a biobased polymer with high resilience and high elongation at break and thus can be used as an impact modifier for PLA without sacrificing sustainability. But NR is also immiscible with PLA. The compatibility of PLA/NR blends was improved by the addition of glycidyl methacrylate-grafted natural rubber (NR-g-GMA), and the impact strength and elongation at break of PLA/NR blend increased about 2.5 times and 2 times, respectively, when 1 wt% NG-g-GMA was introduced, as reported by Punmanee et al.73 Polyamide 610 (PA610) is also a biobased polymer and not miscible with PLA. In order to obtain fully biobased blends with improved mechanical properties, Pai et al.74 compatibilized PLA/PA610 blends with a low molecular weight bisphenol-A type epoxy resin, and found that PA610 could toughen PLA and no-break untorched impact products were obtainable with a suitable content of epoxy resin, due to the formation of copolymers at the interface of the blends via reaction of PLA and PA610 in the presence of the epoxy resin. Shi et al.75 compatibilized blends of PLA and thermoplastic starch (TPS) with addition of GMA-grafted poly(ethylene octane), which is able to react with both PLA and TPS. Wu et al.76 compatibilized a blend of PLA and olefin block copolymer (OBC) with a random terpolymer of ethylene, methyl acrylate and GMA. The compatibilization was evidenced from the reduced particle size of dispersed OBC and the narrowed particle size distribution.
Singh et al.83 enhanced the compatibility of a blend containing major LDPE (80 wt%) and minor PLA (20 wt%) with MA-grafted LDPE. When 4 phr MA-grafted LDPE was added, PLA dispersed uniformly in the LDPE matrix, and optimum mechanical properties were obtained. Yoo et al.84 compatibilized an immiscible blend containing major polypropylene (PP) and minor PLA with PP-g-MAH, and found that the maximum tensile strength and minimum interfacial tension were obtained when 3 phr PP-g-MAH was added for a PP/PLA (80/20, w/w) blend. In a PP/PLA blend consisting of major PLA and minor PP, PP-g-MAH could also be used as a reactive compatibilizer, as reported by Choudhary et al.85 The mechanical properties, especially the toughness, of PP/PLA blends may be improved if ethylene-propylene-diene monomer rubber (EPDM) is added and proper compatibilization occurs for the PP/EPDM/PLA ternary blends. PP-g-MAH alone could not successfully compatibilize the blends in this case, since PP is not miscible with EPDM. While if EPDM-g-MAH was used as a mixing compatibilizer with PP-g-MAH, PP/EPDM/PLA ternary blends with good compatibilization and excellent mechanical properties were obtained, as reported by Park et al.86 Since PP-g-MAH can only react with hydroxyl groups of PLA, the carboxyl groups remained after processing. The compatibilization could be further improved if a co-compatibilizer, which is able to react with carboxyl groups of PLA, was added in the PP/PLA blends. In this regard, Lee et al.87 evaluated the use of PP-g-MAH and PE-g-GMA as a hybrid compatibilizer for PP/PLA blends containing a toughening modifier and compared it with using PP-g-MAH or PE-g-GMA as a single compatibilizer. They found that the hybrid compatibilizer has much better compatibilization efficiency than either PP-g-MAH or PE-g-GMA. In addition, PP-g-MAH was also used as an efficient compatibilizer for PLA/PP/sepiolite nanocomposites as reported by Nunez et al.88 Polycarbonate (PC) is an important engineering plastic with excellent comprehensive performance. To blend with the renewable PLA could endow PC with sustainability. However, PLA and PC are immiscible. In order to improve the compatibility of PLA/PC blends, Lee et al.89 employed three types of maleic anhydride-containing reactive polymers, i.e., poly(styrene-g-acrylonitrile)-maleic anhydride (SAN-g-MAH), poly(ethylene-co-octene) rubber-maleic anhydride (EOR-MAH) and poly(ethylene-co-glycidyl methacrylate) (EGMA), to compatibilize PC/PLA blends. They found that SAN-g-MAH was the most efficient compatibilizer for the blends since the maximum impact and tensile strengths and the minimum interfacial tension were achieved for a PC/PLA (70/30, w/w) blend when 5 phr SAN-g-MAH was added. The best compatibilization efficiency of SAN-g-MAH in PC/PLA blends was ascribed to the partial miscibility between SAN and PC90 and the reaction between MAH and hydroxyl groups of PLA. Teamsinsungvon et al.91 prepared maleic anhydride-grafted PLA (PLA-g-MA) and evaluated the effect of PLA-g-MA on the compatibility of PLA/PBAT blends, and found that the tensile properties of PLA/PBAT with incorporation of PLA-g-MA were much better than those of the uncompatibilized PLA/PBAT blends. Jiang et al.92 also prepared PLA-g-MAH and used it as a compatibilizer in another immiscible blend containing PLA and poly(ethylene terephthalate glycol) (PETG), and found that the phase morphology of the blends became finer with addition of PLA-g-MAH. The optimum content of PLA-g-MAH was 3 phr for a PLA/PETG (80/20, w/w) blend, because it produced the finest phase morphology and the largest elongation at break.
3.4. Addition of low molecular weight chemicals
The main advantage of this method would be the high reactivity of the low molecular weight chemicals, which would react with the blend components quickly during melt blending. The fast reaction makes the method very suitable to be used in the preparation polymer blends through extrusion, which usually occurs in several minutes. One of the challenges of this method is the unmatched viscosity between the low molecular weight chemicals and blend components. It is hard to mix them uniformly due to the rather low viscosity of the low molecular weight chemicals. In addition, the volatility of some low molecular weight chemicals could result in greater harm to operators.
 | ||
| Fig. 7 Chemical structures of isocyanates used in compatibilization of PLA-based blends. | ||
Poly(butylene succinate) (PBS) is another biodegradable polyester can be obtained from renewable resources and shows excellent mechanical properties with good flexibility. It can be used to toughen PLA without sacrificing both sustainability and biodegradability if their compatibility can be improved. Isocyanates should be good reactive compatibilizers for PLA/PBS blends, since the two components contain hydroxyl groups. Harada et al.94 reported the use of LTI to compatibilize PLA/PBS blends during melt extrusion, and found that the impact strength of a PLA/PBS (90/10, w/w) blend was significantly increased from 18 kJ m−2 to 50–70 kJ m−2 when only 0.5 wt% LTI was added, indicating an improvement in compatibility. LTI also succeeded in compatibilizing immiscible blends of PLA and poly(butylene succinate-co-L-lactate) (PBSL) or poly(butylene succinate-co-ε-caprolactone), as evidenced by the changed phase morphologies reported by Vannaladsaysy et al.95,96 Harada et al.97 compared the compatibilization efficiency of four isocyanates, i.e., LTI, LDI, Duranate TPA-100, and Duranate 24A-100, towards immiscible PLLA/PCL blends, and found that the highest impact strength was obtained when LTI was used, but the authors did not explain the reason for the result. The reason might be that the isocyanate group of LTI is more reactive than that of other isocyanates, and thus could react with both blend components sufficiently to compatibilize the blends.
Fang et al.98 compatibilized immiscible blends of PLA and soy protein isolate (SPI) with MDI. The PLA/SPI blends showed a more uniform morphology in the presence of MDI, due to the formation of block or grafted copolymers of PLA and SPI through urethane linkages generated by reaction of isocyanate group of MDI and hydroxyls of blend components. Phetwarotai et al.99,100 investigated the effect of MDI on the properties of PLA/gelatinized starch blends, and found that the interfacial adhesion between the two phases and the tensile properties were improved by addition of 1.25 wt% MDI. Karagoz et al.101 enhanced compatibility and improved mechanical properties of immiscible blends of PLA and citric acid-modified TPS with MDI, both the tensile and impact strengths of the blends being apparently improved by compatibilization with only 1 wt% MDI. MDI was also reported to be efficient in compatibilization of PLA/chitosan blends.102
With the aim of toughening PLA without significant loss in modulus and ultimate tensile strength, Zaman et al.103 blended PLA with a thermoplastic polyester elastomer (TPEE) in the presence of MDI as a reactive compatibilizer, and investigated the effect of MDI content on the mechanical properties and morphologies of PLA/TPEE blends. The results suggested that the dispersed TPEE particle size decreased with increasing MDI content, and the elongation at break of PLA/TPEE (80/20, w/w) increased to more than 200% with only 1 wt% MDI compared to 80% of the control blend, and the tensile strength was also improved slightly by the addition of MDI.
Thermoplastic polyurethane (TPU) with good flexibility can be used to toughen PLA if their immiscibility issue can be resolved. To improve compatibility of immiscible PLA/TPU blends, Dogan et al.104 introduced PDI into PLA/TPU blends during thermal processing, where reactive compatibilization occurred through reactions between isocyanate groups of PDI and hydroxyl/carboxyl groups of PLA and hydroxyl or urethane groups of TPU. The particle size of TPU in a PLA/TPU (80/20, w/w) blend reduced from 1–2 μm to 0.4–1 μm on average with the addition of 1 wt% PDI, and the optimum PDI content for mechanical properties was only 0.5 wt%.
 | ||
| Fig. 8 Proposed reactions in compatibilization of PLA-based blends with free radical initiator. | ||
Wang et al.105 improved compatibility of PLA/PBS blends via an in situ compatibilization procedure in the presence of DCP, and found that when only 0.1 wt% DCP was added, the particle size of dispersed PBS reduced significantly to 0.2–1.0 μm and the blend showed a uniform morphology, which were very beneficial for obtaining excellent mechanical properties. The notched impact strength of a PLA/PBS (80/20, w/w) blend with addition of 0.1 wt% DCP showed the highest value of 30 kJ m−2, compared to 3.7 kJ m−2 for the blend without DCP. Ji et al.106 also studied the effect of DCP on the morphology and properties of PLA/PBS blends, and found that the compatibility of PLA/PBS was increased significantly as evidenced by the decreased dispersed PBS particle size and the finer phase morphology with increasing DCP content, which resulted in significant improvement in mechanical properties. Also, the addition of DCP could cause branched and crosslinked structure, which could work as a nucleation site to accelerate the crystallization rate of PLA. DCP was also applied in compatibilization of PLA/PBAT blends, as reported by Ma et al.107 They found that the introduction of DCP during thermal blending of PLA and PBAT could lead to a reduction in PBAT domain size and an enhancement in interfacial adhesion. The elongation at break and impact strength of the PLA were increased to 300% and 110 J m−1, respectively, after blending with PBAT in the presence of DCP. Branched/crosslinked structure also formed as evidenced by the solid-like behavior in the low-frequency zone. Another peroxide, 2,5-dimethyl-2,5-di(tert-butylperoxy)hexane, was also reported to improve the compatibility of PLA/PBAT blends by Coltelli et al.108 The best compatibilization efficiency was obtained by the addition of 0.2 wt% peroxide for a PLA/PBAT (75/25, w/w) blend, which showed the largest elongation at break and the best phase morphology with smallest PBAT particles dispersed uniformly in PLA matrix. The compatibilization mechanism was proposed as the formation of copolymers consisting of PLA and PBAT through the reaction of macro-radicals PBAT˙ or PBAT–O–O˙ with PLA˙ or PLA–O–O˙ generated in the presence of the peroxide. Dong et al.109 investigated the effect of DCP on the morphology and properties of fully biobased polymer blends of PLA and PHB, and found that the size of dispersed PLA phase was reduced obviously and the phase boundary became unclear on addition of DCP. The mechanical properties of PLA/PHB blends were significantly improved, indicating improved compatibility, which was ascribed to the formation of PHB-g-PLA copolymers and/or PHB-crosslink-PLA network at the interfaces. The compatibility of immiscible PLA/NR blends could be also significantly enhanced by addition of DCP through the formation of crosslinking copolymers containing both PLA and NR, as reported by Huang et al.110
Shin et al.112 compatibilized PLA/PCL blends through addition of GMA monomer with the help of electron-beam irradiation. Two steps were involved in this technique. In the first step, melt blending of PLA, PCL and GMA was carried out in a twin-screw co-rotating extruder, which caused the formation of PLA/PCL blends with GMA located at the interface. The existence of GMA could result in a fine dispersion of minor PCL in the PLA matrix. In the second step, electron-beam irradiation was used to initiate the cross-copolymerization of GMA at the interface to enhance interfacial adhesion between PLA and PCL phases. The combination of the two steps could result in a significant improvement in compatibility between PLA and PCL.
Xiong et al.113 compatibilized immiscible PLA/starch blends with epoxidized soybean oil (ESO), which is usually used as an eco-friendly plasticizer for PVC and chlorinated rubber and contains several epoxy groups available for the reaction with carboxyl and hydroxyl groups of polymers. Limited improvement in compatibility was obtained by direct blending of PLA with starch and ESO, due to the relatively low reactivity of hydroxyl towards epoxy groups of ESO, while sufficient compatibilization occurred if MA-grafted native starch (MGST) was used to replace native starch to blend with PLA and ESO, owning to the increased reaction possibility between ESO and MGAT. The formation of the actual compatibilizer, i.e., copolymer of starch and PLA, is shown in Fig. 9.
 | ||
| Fig. 9 Possible reaction in PLA/MGST blends compatibilized by ESO during melt blending process. Reprinted with permission from ref. 113. Copyright 2014 Elsevier. | ||
Jariyasakoolroj et al.114 reported the compatibilization of PLA and starch by the use of chloropropyltrimethoxysilane (CPMS), which was first grafted onto the surface of the starch through formation of covalent bonds to modify the starch. The CPMS-grafted starch would then react with PLA during blending to generate copolymers of PLA and starch, which provided compatibility between PLA and starch and also worked as a nucleating agent to significantly increase the degree of crystallinity.
3.5. Incorporation of reactive groups on the polymer chains of one component
This approach is similar to addition of reactive polymers with respect to the compatibilization mechanism. Both techniques involve the formation of copolymers through the reaction between the reactive group of a reactive polymer and the terminal/pendent groups of the other component. The difference is that the blending component in this case is the reactive polymer. This technique may have better compatibilization efficiency since there are direct covalent bonds between PLA and blend components through functional group reactions.Epoxy is one of the most widely reported reactive groups incorporated into polymers to prepare compatible PLA blends, which should be ascribed to the high reactivity towards terminal groups of PLA and easy introduction of epoxy to polymers through copolymerization of GMA with other vinyl monomers. In order to improve the compatibility between liquid natural rubber and PLA, Nghia et al.115 modified the liquid natural rubber by incorporation of epoxy groups, which would react with the terminal carboxyl groups of PLA to form covalent bonding at the interface to enhance the interfacial adhesion and improve the compatibility. Oyama116 prepared a super-tough PLA blend by reactive blending of PLA with an ethylene copolymer, EGMA, which contains 3 wt% GMA. The epoxy groups could react with the terminal carboxyl and hydroxyl groups of PLA leading to sufficient compatibility between the two components. Super-tough PLA/EGMA blends with impact strength of 72 kJ m−2 could be obtained when the dispersed EGMA particle size was reduced to 100–300 nm under suitable processing conditions. Su et al.117 prepared compatibilized PLA binary blends by blending with a GMA-grafted poly(ethylene octane) (GMA-g-POE). The good compatibility was also ascribed to the reaction between epoxy groups of GMA-g-POE and terminal groups of PLA. In order to toughen PLA with ABS and improve compatibility of PLA/ABS blends, Su et al.118 prepared GMA-functionalized ABS (ABS-g-GMA) and used it as a component to blend with PLA. They found that compatibilization and crosslinking took place simultaneously between the epoxy groups of ABS-g-GMA and the end carboxyl or hydroxyl groups of PLA. Super toughness of the blend was obtained, as evidenced by an impact strength of 540 J m−1 when only 1 wt% GMA was grafted to ABS. When some other polymers such as poly(butyl acrylate-co-ethyl acrylate) and poly(methyl methacrylate-co-butadiene) were modified by GMA, they also showed good compatibility with PLA, and their blends showed excellent mechanical properties, as reported by Hao et al.119,120
Anhydride groups can be incorporated into polymer chains with the aid of free radicals, and thus are sometimes introduced into the blending component of PLA-based blends to improve compatibility. Orozco et al.121 prepared PLA-g-MAH and used it as a component to blend with starch so as to obtain a PLA/starch blend with improved compatibility and mechanical properties. The compatibilization happened by the reaction between anhydride groups of PLA and the side hydroxyl groups of starch.
3.6. Interchange reactions
When two polyesters are blended in the melt state, several interchange reactions, as shown in Fig. 10, can take place to a certain level that depends on the structures of the polymers, the nature and concentration of exchangeable functional groups, blending time and temperature, and the presence and concentration of catalyst. The level of interchange reaction plays a very important role in determining the properties of the final products. Sometimes, complete interchange reaction with formation of a random copolymer can occur if the blending temperature is high enough, or the blending time is sufficiently long, and/or an efficient catalyst is used. Such a level of interchange reaction is usually not preferable, because the random copolymer loses the beneficial properties of both blend components. What we need is a suitable level of interchange reaction, i.e., to an extent where a suitable amount of block copolymer is formed at the interface of the two immiscible components. Thus, the advantageous properties of both components are kept. To get such a level of interchange reaction, carful choice of blending temperature, residence times in melt and suitable content of catalyst if any is required. | ||
| Fig. 10 Interchange reactions for compatibilization of two polyesters. | ||
Yoon et al.122 studied the effects of blend composition and blending time on the interchange reaction and tensile properties of PLA blends with low and high molecular weight PCL (PLA/LPCL/HPCL), and found that a copolymer of PLA and PCL was formed by the ester interchange reaction at 220 °C for 30–60 minutes, and the tensile strength and modulus of blends increased with increasing HPCL content, while the elongation at break of the blend increased with increasing LPCL content.
Coltelli et al.123 investigated the interchange reaction between PLA and PBAT in a discontinuous mixer with tetrabutyl titanate, Ti(OBu)4, as a catalyst, and found that the dispersed PBAT particle size decreased and interfacial adhesion increased with increasing blending time, and that good compatibility could be obtained when the blending was performed at 200 °C for more than 20 min with a Ti(OBu)4 content of 0.07 wt%. Lin et al.124 carried out interchange esterification between PLA and PBAT at 165–175 °C through melt extrusion at a screw speed of 90 rpm. They studied the effect of the added content of the catalyst Ti(OBu)4 on the compatibility and mechanical properties of PLA/PBAT blends, and found that large PBAT particles dispersed non-uniformly in the PLA matrix in pristine PLA/PBAT blend and a distinct interface can be observed, while when Ti(OBu)4 was added, the size of PBAT particles decreased and the interface became obscure, indicating improved compatibility. The particle size of PBAT was reduced to 0.5 μm when 0.5 wt% Ti(OBu)4 was added and the blends showed tensile strength, elongation at break and impact strength of 45 MPa, 298% and 9 kJ m−2, respectively, compared to 35 MPa, 46% and 5 kJ m−2 for the pristine PLA/PBAT blend.
Sadik et al.125 studied the interchange reaction between PLA and poly(ethylene-co-vinylalcohol) (EVOH) in the absence and presence of different catalysts, i.e., 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and tin(II) bis(2-ethylhexanoate) (Sn(Oct)2). The reaction was accelerated by the addition of both catalysts, but Sn(Oct)2 showed higher catalytic efficiency than TBD. The reaction finished in only a few minutes if Sn(Oct)2 was used as the catalyst at 200 °C. PLA-grafted EVOH was formed through the reaction as confirmed by thermal, thermomechanical and 1H NMR analyses.
To improve compatibility and physical properties of PLA blends with PC, Phuong et al.126 added tetrabutylammonium tetraphenylborate (TBATPB) and triacetin (TA) into the blends during melt processing. Interchange reaction between PLA and PC occurred when TBATPB and TA were used as catalysts, and thus PLA–PC copolymers were formed in the blends. Consequently, the compatibility between PLA and PC was improved significantly with co-continuous phase morphology formed, and mechanical strength of the compatibilized blends was improved significantly.
Recently, Thurber et al.127 improved the compatibility of PLA/PE blends by addition of interfacially localized catalysts. In the study, the compatibilization occurred through interchange reaction between telechelic hydroxyl functional PE and PLA. Interfacially localized catalyst (stannous octoate) accelerated the interchange reaction and resulted in improved compatibility. The size of dispersed PLA domain decreased while the number of dispersed PLA particles increased considerably.
Interchange reaction is a simple, efficient, and eco-friendly method to compatibilize PLA-based blends. However, the method has some limitations, as it is only applicable when the blend components have exchangeable functional groups. Such reaction could not happen between PLA and some vinyl polymers. In addition, interchange reaction usually occurs at high temperatures, where thermal degradation of the components cannot be avoided, which would lead to deterioration of properties, and undesirable color and appearance.
3.7. Dynamic vulcanization and interfacial compatibilization
Dynamic vulcanization involves a process of selective crosslinking of a rubber phase during melt blending with a thermoplastic polymer, resulting in a two-phase material in which the crosslinked rubber phase is dispersed in the thermoplastic matrix. Dynamic vulcanization could prevent the dispersed rubber phase from undergoing coalescence, thus affecting the final morphology and properties of the resulting materials. When two polymers with similar polarities are blended, a fine morphology can be frozen through dynamic vulcanization and no extra compatibilizer is required. But dynamic vulcanization of two incompatible polymers usually leads to coarse morphology, where in situ compatibilization is still required to increase the interfacial adhesion, and then a blend with high performance is obtainable. The process involving both dynamic vulcanization and in situ compatibilization is referred to as dynamic vulcanization and interfacial compatibilization. This technique is very useful to compatibilize PLA blends with curable elastic polymers with the aim of improving toughness. The main advantage of this technique is the ability to control the phase morphology and therefore the properties of the resulting blends through the extent of vulcanization and interfacial reaction. This method is usually limited to blends which contain curable polymers, but cannot be applicable in other systems without curable polymers.The dispersed phase in this technique is a rubber, which is a very efficient toughening agent for brittle polymers. Therefore, this technique is very powerful for toughening of PLA. Liu et al.128–130 for the first time employed this technique to prepare PLA-based materials with super toughness. The rubber phase that they used is an ethylene-butyl acrylate-glycidyl methacrylate (EBA-GMA) terpolymer, and a zinc ionomer of ethylene methyacrylic acid copolymer (EMAA-Zn) was used as a catalyst, of which the carboxyl group initiated the crosslinking of epoxy groups in the EBA-GMA phase, and zinc ions catalyzed the reaction between the epoxy groups of EBA-GMA and the terminal groups of PLA to improve interfacial adhesion between the dispersed rubber phase and the PLA matrix. Morphological analysis indicated that the binary blend of PLA/EBA-GMA (80/20, w/w) showed very fine morphology with EBA-GMA dispersed uniformly in the PLA matrix at the size of ∼0.3 μm due to the good compatibility of the blend resulting from the reaction of epoxy of EBA-GMA and hydroxyl of PLA; the vulcanization of EBA-GMA after introduction of EMAA-Zn led to an increase in the size of dispersed phase in the PLA/EBA-GMA/EMAA-Zn (80/10/10, w/w/w) to ∼0.83 μm, which is in the optimum range for high toughening efficiency in rubber-toughened PLLA blends. Through dynamic vulcanization and interfacial compatibilization, the ternary blends showed elongation at break and notched impact strength of 229.1% and 777.2 J m−1, respectively, compared to 10.2% and 101.9 J m−1 for the binary PLA/EBA-GMA blend.
Chen et al.131 employed dynamic vulcanization and interfacial compatibilization to improve compatibility between PLA and NR by melt blending in the presence of DCP as an initiator. The thermal decomposition of DCP led to the formation of free radicals, which then initiated vulcanization of NR and also caused the formation of PLA macroradicals that reacted with NR to generate copolymers to enhance interfacial adhesion between PLA and vulcanized NR. A super-tough blend with notched impact strength of 58.3 kJ m−2 was obtained when 35 wt% NR was blended with 65 wt% PLA. It is interesting that in such a system, the minor phase of NR changed to the continuous phase while the major PLA phase became the dispersed phase after dynamic vulcanization and interfacial compatibilization. But the mechanism for the formation of continuous vulcanized NR phase was unclear.
Fang et al.132 reported super-tough PLA blends with poly(ethylene glycol) diacylate (PEGDA) monomer via dynamic vulcanization and interfacial compatibilization without additional radical initiators. Rheological measurement suggested that thermally induced crosslinking of PEGDA occurred when the temperature was increased to higher than 116 °C in the absence of additional initiator. They thought that the polymerization may be initiated by free radical species such as radical impurities, peroxides, and oxygen plasma. When PEGDA was melt blended with PLA, the crosslinking of PEGDA finished within the blending period of 10 min, as evidenced by the change of melt torque of the blend. FT-IR analysis indicated that in situ compatibilization took place by interchange esterification of PLA and PEGDA during melt blending. The blends after dynamic vulcanization and interfacial compatibilization showed notched impact strength up to 50 kJ m−2 when 15 wt% PEGDA was introduced.
Recently, we have introduced two different crosslinked polyurethanes (CPUs), which were obtained via in situ polymerization, as the rubber phases to toughen PLA through dynamic vulcanization and interfacial compatibilization.133,134 In one case, the CPU was composed of poly(ethylene glycol) (PEG) and polymeric methylenediphenylene diisocyanate (PMDI). During blend preparation, PLA was first premixed with PEG in an internal mixer at 190 °C for 4 min, the blending being finished when the melt torque leveled off after the addition of PMDI. The CPU was in situ formed through polymerization of PEG with PMDI, and interfacial compatibilization occurred by the reaction of isocyanate groups with the terminal groups of PLA, as evidenced by FT-IR analysis. A super-tough PLA blend was obtained when the content of the incorporated CPU was 30% with elongation at break and notched impact strength of ∼250% and 546 J m−1, respectively. In order to further investigate the effect of crosslinking density of CPU on the properties of CPU-toughened PLA blends, PMDI was replaced by MDI and glycerol in the other system. The crosslinking density, which was proved to be very important in determining phase morphology and mechanical properties of PLA/CPU blends, could be tuned by the content of the tri-functional monomer glycerol. The particle size of CPU in PLA matrix increased gradually while the notched impact strength increased first and then decreased with increasing CPU crosslinking density. The highest notched impact strength of 407.6 J m−1 was achieved for a PLA/CPU (80/20, w/w) blend that contained 10 wt% glycerol (based on PEG weight), and the particle size of CPU in this blend was ∼0.76 μm, which was just in the optimum range for rubber-toughened PLA blends.
In order to produce super-tough PLA materials without compromising sustainability, we prepared an unsaturated aliphatic polyester elastomer (UPE) from biobased monomers and melt blended it with PLA in the presence of DCP as a free radical initiator via a dynamic vulcanization and interfacial compatibilization technique.135 During melt processing, dynamic vulcanization of UPE occurred by the initiation of free radicals formed via thermal decomposition of DCP. Meanwhile free radicals could abstract hydrogen from PLA polymer chains to generate PLA macroradicals, which then grafted onto the vulcanized UPE dispersed phases via attacking the double bonds of the UPE to form grafting copolymers, which located at the interface to significantly improve the interfacial adhesion between the two phases. Fig. 11 shows the proposed reactions for the preparation of PLA blends with UPE through dynamic vulcanization and interfacial compatibilization. The blend was named thermoplastic vulcanizate (TPV) which refers to a polymer blend with a crosslinked rubber phase dispersed finely in a thermoplastic matrix. Super-tough TPV with elongation at break and notched impact strength of 259.9% and 586.6 J m−1 was obtained by blending PLA with 20 wt% UPE in the presence of 0.2 phr DCP at 180 °C and 50 rpm for about 10 min.
 | ||
| Fig. 11 Possible reactions in preparation of super-tough PLA blends with unsaturated aliphatic polyester elastomer (UPE) through dynamic vulcanization and interfacial compatibilization. Reprinted with permission from ref. 135. Copyright 2014 American Chemical Society. | ||
3.8. Addition of nanoparticles
Addition of nanoparticles to an immiscible blend provides an alternative way of improving compatibility of the blend. Nanoparticles usually locate at the interface of the components, acting as interfacial modifiers to strengthen the interfacial adhesion and therefore the performance of the blend. Nanoparticles can prevent preformed fine dispersed phase particles from coalescence during melt processing, thus stabilizing the fine morphology and therefore maintaining the properties of the blend. Unlike polymer compatibilizers, nanoparticles are unspecific to the nature of immiscible blend components, and easily incorporated via blending. In addition to improving compatibility, the incorporation of nanoparticles can generate high-performance materials that combine the advantages of polymer blends and the merits of polymer nanocomposites. Therefore, tailoring phase morphology of immiscible blends by addition of nanoparticles represents a universal way of preparing compatible polymer blend nanocomposites with improved physical properties.Chen et al.47 evaluated the effect of twice-functionalized organoclay (TFC) on the compatibility between PLA and PBS, and found that the content of TFC played an important role in the morphology of the blend. TFC exfoliated fully and dispersed almost exclusively in the PLA matrix and the domain size of dispersed PBS did not change considerably when the content of TFC was less than 0.5 wt%; while as the TFC content increased, the clay layers dispersed in both PLA and PBS phases and the domain size of PBS became much smaller and increased gradually with further increasing TFC content. This change with increasing TFC content was attributed to two factors. On the one hand, TFC with functional groups could react with both PLA and PBS, thus acting like a reactive compatibilizer. On the other hand, some clay layers that located at the interface of PLA/PBS blend could prevent coalescence of the dispersed domains and contribute to the reduction in the domain size. Hoidy et al.136 compatibilized PLA/PCL blends with organoclay (OMMT), and found that the presence of OMMT as a filler not only enhanced the dispersion and interfacial adhesion of the polymer matrix but also improved mechanical properties and thermal stability of PLA/PCL blends. Ojijo et al.137 compatibilized PLA/PBSA blends with organoclay (C20A) and investigated the effect of the content of C20A on the properties of the blends. They found that the optimum properties for a PLA/PBSA (70/30, w/w) blend was obtained when the content of C20A was 2 wt%. Risse et al.138 tailored the morphology and properties of PLA/PBS blend by addition of an organoclay (Cl30B). They found that Cl30B exfoliated partially in the blend and its content played an important role in the morphology and properties of the blend. Co-continuous morphology of a PLA/PBS (50/50, w/w) blend with a ductile behavior was achieved when 3 wt% Cl30B was added, while lamellar morphology accompanied by brittleness was obtained with further increasing Cl30B content. A recent study by Ferreira et al.139 suggested that the addition of Cloisite® 30B (C30B) into TPS/PLA blends would improve adhesion between the two phases, compared to the unmodified binary blends.
Carbon nanotubes are a new type of anisotropic one-dimensional nanoparticles, which have attracted considerable attention in terms of reinforcing physical properties of polymer matrices through nanocomposite technology, due to their extraordinarily high elastic modulus, strength, and resilience. They could also be used in improving the compatibility of immiscible blends via selective localization, as reported by Wu et al.140 They functionalized multiwalled carbon nanotubes (MWCNT) with carboxyl groups, and added the functionalized MWCNT into melts of immiscible PCL/PLA blend during mixing with a rheometer. The compatibility between PCL and PLA was improved obviously by the addition of functionalized MWCNT, as evidenced by the significant reduction of dispersed PLA domain size and the enhanced interfacial adhesion. The dispersed PLA particle size in a PCL/PLA (70/30, w/w) blend decreased apparently from 21.5 to 6.3 μm as the MWCNT loadings increased up to 1 phr. The improved compatibility was attributed to the special morphological structure that was formed in which carboxylic MWCNTs mainly dispersed in the PCL matrix and at the phase interface. The compatibilized PCL/PLA blends then showed highly improved performance with respect to rheological, conductive, and mechanical properties as compared with those of unmodified PCL/PLA blend.
Another nanoparticle that has been used to compatibilize immiscible PLA blends is silica, as reported by Odent et al.141 in PLA blend with a rubbery ε-caprolactone-based copolyester, P[CL-co-LA]. They found that P[CL-co-LA] with sphere-like nodules dispersed regularly in the PLA matrix in a blank blend containing 10 wt% P[CL-co-LA], while the spherical nodules disappeared when 5 wt% hexamethyldisilazane surface-treated fumed silica nanoparticles were added and oblong microstructures began to appear, and more interestingly co-continuous morphology was observed with further increasing silica nanoparticle content up to 10 wt%, as shown in Fig. 12. Such a morphological change was ascribed to the presence of large-surface silica nanoparticles at the interface of the blend components. The impact strength of the blend by addition of silica was significantly increased from 11.4 kJ m−2 for the blank blend to 27.3 and 39.7 kJ m−2 for blends containing 5 and 10 wt% silica, respectively.
 | ||
| Fig. 12 TEM images of room-temperature notched surfaces of PLA-based materials containing 10 wt% of P[CL-co-LA] copolyester without silica nanoparticles (a), and with 5 wt% (b and b′) and 10 wt% of silica nanoparticles (c and c′). Reprinted with permission from ref. 141. Copyright 2013 Elsevier. | ||
Recently, Monticelli et al.142 evaluated the effects of different types of modified polyhedral oligomeric silsesquioxane (POSS) on the compatibility of PLA/PCL blends. Addition of unmodified POSS reduced the size of PCL domains; addition of hydroxyl group-functionalized POSS (POSS–OH) increased the adhesion between PLA and PCL; and addition of poly(ε-caprolactone)-b-poly(L-lactide) diblock copolymer-grafted POSS (POSS-PCL-b-PLLA) led to the formation of an almost homogeneous microstructure. All the characteristics indicated the improved compatibility of the blends.
3.9. Other strategies
Some other techniques have been sporadically reported for the compatibilization of some specific immiscible PLA blends. Addition of a third polymer that is miscible with both PLA and the other component could improve compatibility of PLA-based blends. But due to the lack of such polymers, this technique can only be used in very special blend pairs. Poly(vinyl acetate) is miscible with both PLA and PHB, and thus can be used to improve compatibility of PLA/PHB blends.143 Poly(ethylene oxide) (PEO) is miscible with PLA and PCL and the presence of PEO in a PLA/PCL blend could result in a single-phase completely miscible blend under suitable conditions, as reported by Buddhiranon et al.144Introducing some special interactions such as hydrogen bonding between PLA and the other blend component is also able to improve compatibility. The work by Kuo et al.145 indicated that addition of bisphenol A (BPA) was able to improve compatibility between PLA and PCL since both polymers are miscible with BPA. In order to improve compatibility between PLA and polystyrene (PS), Zuza et al.44 incorporated hydroxyl groups into PS by copolymerization of styrene with hydroxystyrene (HS) and then blended the modified PS with PLA. The introduction of hydroxyl groups to PS led to the formation of hydrogen bonding with carbonyl groups of PLA, resulting in significant improvement in compatibility between the two polymers. A completely miscible blend was even achieved when the molar content of HS was increased to 16%.
4. Conclusions
Polymer blending provides a simple and economic way to modify properties and thus expand applications of PLA. In this sense, a variety of polymers have been blended with PLA. Unfortunately, most of the polymers are thermodynamically immiscible with PLA. High-performance blends are unanticipated without compatibilization. Various compatibilization strategies, including addition of copolymers, addition of reactive polymers, addition of low molecular weight chemicals, addition of nanoparticles, interchange reactions, dynamic vulcanization and interfacial compatibilization, etc., have been applied to improve compatibility between PLA and different blend components. The addition of copolymers is the conventional and efficient way to compatibilize polymer blends. This technique shows very good compatibilization efficiency. However, a drawback is the commercial unavailability of the specific copolymers, which have to be synthesized prior to blending. This method seems undesirable especially for industrial application due to the additional investment regarding preparation of the specific copolymers. Interchange reaction represents an economic way by melt blending PLA and other polymers with exchangeable functional groups. But thermal degradation usually occurs during interchange reaction, which would cause a deterioration of properties combined with undesirable color and appearance. The other techniques such as addition of reactive substances and dynamic vulcanization and interfacial compatibilization involving chemical reactions between PLA and blend components or compatibilizers can be called reactive compatibilizations, which are much more applicable not only for laboratory study but also for industrial application, due to the availability of various reactive substances and high compatibilization efficiency of the techniques. In addition, the introduction of nanoparticles to modify the interfacial properties and phase morphologies should be a very promising technique to compatibilize PLA-based blends, as the presence of nanoparticles would not only improve the compatibility between PLA and blend components, but also reinforce the mechanical strength or impart some novel functionalities to the resulting composites. Therefore, more efforts should focus on the compatibilization of PLA-based blends with various nanoparticles and thus to develop novel properties and expand applications of PLA-based materials.Acknowledgements
This work was supported by National Natural Science Foundation of China (20904034) and Fundamental Research Funds for the Central Universities (XDJK2015C022).References
- M. M. Reddy, S. Vivekanandhan, M. Misra, S. K. Bhatia and A. K. Mohanty, Prog. Polym. Sci., 2013, 38, 1653–1689 CrossRef CAS PubMed.
- L. Yu, K. Dean and L. Li, Prog. Polym. Sci., 2006, 31, 576–602 CrossRef CAS PubMed.
- R. Chandra and R. Rustgi, Prog. Polym. Sci., 1998, 23, 1275–1335 CrossRef.
- D. Garlotta, J. Polym. Environ., 2001, 9, 63–84 CrossRef CAS.
- M. H. Hartmann, High Molecular Weight Polylactic Acid Polymers, in Biopolymers from Renewable Resources, Springer-Verlag, Berlin, 1998, ch. 15, pp. 367–411 Search PubMed.
- R. Datta and M. Henry, J. Chem. Technol. Biotechnol., 2006, 81, 1119–1129 CrossRef CAS.
- W. H. Carothers, G. L. Dorough and F. J. van Natta, J. Am. Chem. Soc., 1932, 54, 761–772 CrossRef CAS.
- R. M. Rasal, A. V. Janorkar and D. E. Hirt, Prog. Polym. Sci., 2010, 35, 338–356 CrossRef CAS PubMed.
- J. Tuominen and J. V. Seppala, Macromolecules, 2000, 33, 3530–3535 CrossRef CAS.
- J. B. Zeng, Y. D. Li, W. D. Li, K. K. Yang, X. L. Wang and Y. Z. Wang, Ind. Eng. Chem. Res., 2009, 48, 1706–1711 CrossRef CAS.
- J. B. Zeng, Y. D. Li, Q. Y. Zhu, K. K. Yang, X. L. Wang and Y. Z. Wang, Polymer, 2009, 50, 1178–1186 CrossRef CAS PubMed.
- S. Dutkiewicz, D. Grochowska-Łapienis and W. Tomaszewski, Fibres Text. East. Eur., 2003, 11, 66–70 CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 835–864 CrossRef CAS PubMed.
- J. Ren, N. Zhang and Q. Wang, CN1718607A, 2006.
- T. Maharana, B. Mohanty and Y. S. Negi, Prog. Polym. Sci., 2009, 34, 99–124 CrossRef CAS PubMed.
- Y. Ikada, H. Jamshidi, H. Tsuji and S. H. Hyon, Macromolecules, 1987, 20, 904–906 CrossRef CAS.
- H. Tsuji, F. Horii, S. Hyon and Y. Ikada, Macromolecules, 1991, 24, 2719–2724 CrossRef CAS.
- V. Piemonte, S. Sabatini and F. Gironi, J. Polym. Environ., 2013, 21, 640–647 CrossRef CAS.
- A. Plichta, P. Lisowska, A. Kundys, A. Zychewicz, M. Debowski and Z. Florjanczyk, Polym. Degrad. Stab., 2014, 108, 288–296 CrossRef CAS PubMed.
- J. R. Dorgan, H. J. Lehermeier, L. I. Palade and J. Cicero, Macromol. Symp., 2001, 175, 55–66 CrossRef CAS.
- R. Pantani and A. Sorrentino, Polym. Degrad. Stab., 2013, 98, 1089–1096 CrossRef CAS PubMed.
- G. Gorrasi and R. Pantani, Polym. Degrad. Stab., 2013, 98, 1006–1014 CrossRef CAS PubMed.
- L. T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS PubMed.
- P. J. Jandas, S. Mohanty and S. K. Nayak, Ind. Eng. Chem. Res., 2013, 52, 17714–17724 CrossRef CAS.
- C. J. Weber, V. Haugaard, R. Festersen and G. Bertelsen, Food Addit. Contam., 2002, 19, 172–177 CrossRef CAS PubMed.
- Y. Ikada and H. Tsuji, Macromol. Rapid Commun., 2000, 21, 117–132 CrossRef CAS.
- Y. Wang, S. M. Chiao, T. F. Hung and S. Y. Yang, J. Appl. Polym. Sci., 2012, 125, E402–E412 CrossRef CAS.
- K. S. Anderson, K. M. Schreck and M. A. Hillmyer, Polym. Rev., 2008, 48, 85–108 CrossRef CAS.
- H. Liu and J. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1051–1083 CrossRef CAS.
- J. M. Raquez, Y. Habibi, M. Murariu and P. Dubois, Prog. Polym. Sci., 2013, 38, 1504–1542 CrossRef CAS PubMed.
- C. Zeng, N. W. Zhang and J. Ren, J. Appl. Polym. Sci., 2012, 125, 2564–2576 CrossRef CAS.
- R. Slivniak and A. J. Domb, Macromolecules, 2005, 38, 5545–5553 CrossRef CAS.
- D. Haynes, N. K. Abayasinghe, G. M. Harrison, K. J. Burg and D. W. Smith Jr, Biomacromolecules, 2007, 8, 1131–1137 CrossRef CAS PubMed.
- A. Meduri, T. Fuoco, M. Lamberti, C. Pellecchia and D. Pappalardo, Macromolecules, 2014, 47, 534–543 CrossRef CAS.
- B. M. Chamberlain, B. A. Jazdzewski, M. Pink, M. A. Hillmyer and W. B. Tolman, Macromolecules, 2000, 33, 3970–3977 CrossRef CAS.
- W. D. Li, J. B. Zeng, Y. D. Li, X. L. Wang and Y. Z. Wang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5898–5907 CrossRef CAS.
- N. Roy, R. Sengupta and A. K. Bhowmick, Prog. Polym. Sci., 2012, 37, 781–819 CrossRef CAS PubMed.
- D. Cohn and A. H. Salomon, Biomaterials, 2005, 26, 2297–2305 CrossRef CAS PubMed.
- J. B. Zeng, Y. D. Li, Y. S. He, S. L. Li and Y. Z. Wang, Ind. Eng. Chem. Res., 2011, 50, 6124–6131 CrossRef CAS.
- Y. S. He, J. B. Zeng, S. L. Li and Y. Z. Wang, Thermochim. Acta, 2012, 529, 80–86 CrossRef CAS PubMed.
- C. Koning, M. V. Duin, C. Pagnoulle and R. Jerome, Prog. Polym. Sci., 1998, 23, 707–757 CrossRef CAS.
- Y. B. Wang and M. A. Hillmyer, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2755–2766 CrossRef CAS.
- Y. Li and H. Shimizu, Eur. Polym. J., 2009, 45, 738–746 CrossRef CAS PubMed.
- E. Zuza, A. Lejardi, J. M. Ugartemendia, N. Monasterio, E. Meaurio and J. R. Sarasua, Macromol. Chem. Phys., 2008, 209, 2423–2433 CrossRef CAS.
- C. S. Wu, Macromol. Biosci., 2005, 5, 352–361 CrossRef CAS PubMed.
- J. Qiu, C. Xing, X. Cao, H. Wang, L. Wang, L. Zhao and Y. Li, Macromolecules, 2013, 46, 5806–5814 CrossRef CAS.
- G. X. Chen, H. S. Kim, E. S. Kim and J. S. Yoon, Polymer, 2005, 46, 11829–11836 CrossRef CAS PubMed.
- Y. H. Na, Y. He, X. Shuai, Y. Kikkawa, Y. Doi and Y. Inoue, Biomacromolecules, 2002, 3, 1179–1186 CrossRef CAS PubMed.
- D. R. Paul and J. W. Barlow, Polym. Rev., 1980, 18, 109–168 Search PubMed.
- B. Imre and B. Pukanszky, Eur. Polym. J., 2013, 49, 1215–1233 CrossRef CAS PubMed.
- C. H. Kim, K. Y. Cho, E. J. Choi and J. K. Park, J. Appl. Polym. Sci., 2000, 77, 226–231 CrossRef CAS.
- H. Tsuji, T. Yamada, M. Suzuki and S. Itsuno, Polym. Int., 2003, 52, 269–275 CrossRef CAS.
- N. S. Choi, C. H. Kim, K. Y. Cho and J. K. Park, J. Appl. Polym. Sci., 2002, 86, 1892–1898 CrossRef CAS.
- W. Bai, Q. Li, L. Y. Jiang, Z. P. Zhang, S. L. Zhang and C. D. Xiong, J. Appl. Polym. Sci., 2011, 120, 544–551 CrossRef CAS.
- X. Xie, W. Bai, A. Wu, D. Chen, C. Xiong, C. Tang and X. Pang, J. Appl. Polym. Sci., 2015, 132, 41323 Search PubMed.
- Z. Ma, N. Zhao and C. Xiong, Bull. Mater. Sci., 2012, 35, 575–578 CrossRef CAS PubMed.
- K. S. Anderson, S. H. Lim and M. A. Hillmyer, J. Appl. Polym. Sci., 2003, 89, 3757–3768 CrossRef CAS.
- R. DellErba, G. Groeninckx, G. Maglio, M. Malinconico and A. Migliozzi, Polymer, 2001, 42, 7831–7840 CrossRef CAS.
- G. Maglio, A. Migliozzi, R. Palumbo, B. Immirzi and M. G. Volpe, Macromol. Rapid Commun., 1999, 20, 236–238 CrossRef CAS.
- G. Maglio, M. Malinconico, A. Migliozzi and G. Groeninckx, Macromol. Chem. Phys., 2004, 250, 946–950 CrossRef.
- D. Wu, Y. Zhang, L. Yuan, M. Zhang and W. Zhou, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 756–765 CrossRef CAS.
- W. Chumeka, P. Pasetto, J. F. Pilard and V. Tanrattanakul, J. Appl. Polym. Sci., 2015, 132, 41426 CrossRef.
- K. Chang, M. L. Robertson and M. A. Hillmyer, ACS Appl. Mater. Interfaces, 2009, 1, 2390–2399 CAS.
- V. Vilay, M. Mariatti, Z. Ahmad, K. Pasomsouk and M. Todo, Mater. Sci. Eng., A, 2010, 527, 6930–6937 CrossRef PubMed.
- V. Vilay, M. Mariatti, Z. Ahmad, K. Pasomsouk and M. Todo, Polym. Adv. Technol., 2011, 22, 1786–1793 CrossRef CAS.
- X. Yang, A. Finne-Wistrand and M. Hakkarainen, Compos. Sci. Technol., 2013, 86, 149–165 CrossRef CAS PubMed.
- J. Wootthikanokkhan, P. Kasemwananimit, N. Sombatsompop, A. Kositchaiyong, S. I. N. Ayutthaya and N. Kaabbuathong, J. Appl. Polym. Sci., 2012, 126, E388–E395 CrossRef CAS.
- N. Zhang, Q. Wang, J. Ren and L. Wang, J. Mater. Sci., 2009, 44, 250–256 CrossRef CAS PubMed.
- Y. F. Kim, C. N. Choi, Y. D. Kim, K. Y. Lee and M. S. Lee, Fibers Polym., 2004, 5, 270–274 CrossRef CAS.
- S. M. Lai, K. C. Hung, H. C. Kao, L. C. Liu and X. F. Wang, J. Appl. Polym. Sci., 2013, 130, 2399–2409 CrossRef CAS.
- M. Y. Jo, Y. J. Ryu, J. H. Ko and J. S. Yoon, J. Appl. Polym. Sci., 2012, 125, E231–E238 CrossRef CAS.
- R. Al-Itry, K. Lamnawar and A. Maazouz, Polym. Degrad. Stab., 2012, 97, 1898–1914 CrossRef CAS PubMed.
- P. Juntuek, C. Ruksakulpiwat, P. Chumsamrong and Y. Ruksakulpiwat, J. Appl. Polym. Sci., 2012, 125, 745–754 CrossRef CAS.
- F. C. Pai, S. M. Lai and H. H. Chu, J. Appl. Polym. Sci., 2013, 130, 2563–2571 CrossRef CAS.
- Q. Shi, C. Chen, L. Gao, L. Jiao, H. Xu and W. Guo, Polym. Degrad. Stab., 2011, 96, 175–182 CrossRef CAS PubMed.
- M. Wu, Z. Wu, K. Wang, Q. Zhang and Q. Fu, Polymer, 2014, 55, 6409–6417 CrossRef CAS PubMed.
- R. Bhardwaj and A. K. Mohanty, Biomacromolecules, 2007, 8, 2476–2484 CrossRef CAS PubMed.
- M. A. Huneault and H. Li, Polymer, 2007, 48, 270–280 CrossRef CAS PubMed.
- J. F. Zhang and X. Z. Sun, Biomacromolecules, 2004, 5, 1446–1451 CrossRef CAS PubMed.
- S. W. Hwang, J. K. Shim, S. Selke, H. Soto-Valdez, M. Rubino and R. Auras, Macromol. Mater. Eng., 2013, 298, 624–633 CrossRef CAS.
- N. Wang, J. Yu and X. Ma, Polym. Int., 2007, 56, 1440–1447 CrossRef CAS.
- P. J. Jandas, S. Mohanty and S. K. Nayak, ACS Sustainable Chem. Eng., 2014, 2, 377–386 CrossRef CAS.
- G. Singh, H. Bhunia, A. Rajor, R. N. Jana and V. Choudhary, J. Appl. Polym. Sci., 2010, 118, 496–502 CrossRef CAS.
- T. W. Yoo, H. G. Yoon, S. J. Choi, M. S. Kim, Y. H. Kim and W. N. Kim, Macromol. Res., 2010, 18, 583–588 CrossRef CAS.
- P. Choudhary, S. Mohanty, S. K. Nayak and L. Unnikrishnan, J. Appl. Polym. Sci., 2011, 121, 3223–3237 CrossRef CAS.
- D. H. Park, M. S. Kim, J. H. Yang, D. J. Lee, K. N. Kim, B. K. Hong and W. N. Kim, Macromol. Res., 2011, 19, 105–112 CrossRef CAS PubMed.
- H. S. Lee and J. D. Kim, Polym. Compos., 2012, 33, 1154–1161 CrossRef CAS.
- K. Nunez, C. Rosales, R. Perera, N. Villarreal and J. M. Pastor, Polym. Bull., 2011, 67, 1991–2016 CrossRef CAS.
- J. B. Lee, Y. K. Lee, G. D. Choi, S. W. Na, T. S. Park and W. N. Kim, Polym. Degrad. Stab., 2011, 96, 553–560 CrossRef CAS PubMed.
- A. Iwan and A. Chuchmała, Prog. Polym. Sci., 2012, 37, 1805–1828 CrossRef CAS PubMed.
- A. Teamsinsungvon, Y. Ruksakulpiwat and K. Jarukumjorn, Polym.-Plast. Technol. Eng., 2013, 52, 1362–1367 CrossRef CAS.
- W. R. Jiang, R. Y. Bao, W. Yang, Z. Y. Liu, B. H. Xie and M. B. Yang, Mater. Des., 2014, 59, 524–531 CrossRef CAS PubMed.
- B. Liu, L. Jiang, H. Liu and J. Zhang, Ind. Eng. Chem. Res., 2010, 49, 6399–6406 CrossRef CAS.
- M. Harada, T. Ohya, K. Iida, H. Hayashi, K. Hirano and H. Fukudal, J. Appl. Polym. Sci., 2007, 106, 1813–1820 CrossRef CAS.
- V. Vannaladsaysy, M. Todo, T. Takayama, M. Jaafar, Z. Ahmad and K. Pasomsouk, J. Mater. Sci., 2009, 44, 3006–3009 CrossRef CAS.
- V. Vannaladsaysy, M. Todo, M. Jaafar, Z. Ahmad and K. Pasomsouk, Polym. Eng. Sci., 2010, 50, 1485–1491 CAS.
- M. Harada, K. Iida, K. Okamoto, H. Hayashi and K. Hirano, Polym. Eng. Sci., 2008, 48, 1359–1368 CAS.
- K. Fang, B. Wang, K. Sheng and X. S. Sun, J. Appl. Polym. Sci., 2009, 114, 754–759 CrossRef CAS.
- W. Phetwarotai, P. Potiyaraj and D. Aht-Ong, J. Appl. Polym. Sci., 2010, 116, 2305–2311 CAS.
- W. Phetwarotai, P. Potiyaraj and D. Aht-Ong, J. Appl. Polym. Sci., 2012, 126, E162–E172 CrossRef CAS.
- S. Karagoz and G. Ozkoc, Polym. Eng. Sci., 2013, 53, 2183–2193 CAS.
- N. E. Suyatma, A. Copinet, V. Coma and F. Fricoteaux, J. Appl. Polym. Sci., 2010, 117, 3083–3091 CAS.
- H. U. Zaman, J. C. Song, L. S. Park, I. K. Kang, S. Y. Park, G. Kwak, B. S. Park and K. B. Yoon, Polym. Bull., 2011, 67, 187–198 CrossRef CAS.
- S. K. Dogan, E. A. Reyes, S. Rastogi and G. Ozkoc, J. Appl. Polym. Sci., 2014, 131, 40251 CrossRef.
- R. Wang, S. Wang, Y. Zhang, C. Wan and P. Ma, Polym. Eng. Sci., 2009, 49, 26–33 CAS.
- D. Ji, Z. Liu, X. Lan, F. Wu, B. Xie and M. Yang, J. Appl. Polym. Sci., 2014, 131, 39580 Search PubMed.
- P. Ma, X. Cai, Y. Zhang, S. Wang, W. Dong, M. Chen and P. J. Lemstra, Polym. Degrad. Stab., 2014, 102, 145–151 CrossRef CAS PubMed.
- M. B. Coltelli, S. Bronco and C. Chinea, Polym. Degrad. Stab., 2010, 95, 332–341 CrossRef CAS PubMed.
- W. Dong, P. Ma, S. Wang, M. Chen, X. Cai and Y. Zhang, Polym. Degrad. Stab., 2013, 98, 1549–1555 CrossRef CAS PubMed.
- Y. Huang, C. Zhang, Y. Pan, W. Wang, L. Jiang and Y. Dan, J. Polym. Environ., 2013, 21, 375–387 CrossRef CAS PubMed.
- W. Dong, B. Zou, P. Ma, W. Liu, X. Zhou, D. Shi, Z. Ni and M. Chen, Polym. Int., 2013, 62, 1783–1790 CrossRef CAS.
- B. Y. Shin and D. H. Han, Radiat. Phys. Chem., 2013, 83, 98–104 CrossRef CAS PubMed.
- Z. Xiong, Y. Yang, J. Feng, X. Zhang, C. Zhang, Z. Tang and J. Zhu, Carbohydr. Polym., 2013, 92, 810–816 CrossRef CAS PubMed.
- P. Jariyasakoolroj and S. Chirachanchai, Carbohydr. Polym., 2014, 106, 255–263 CrossRef CAS PubMed.
- N. Phan Trung, N. Siripitakchai, W. Klinklai, T. Saito, Y. Yamamoto and S. Kawahara, J. Appl. Polym. Sci., 2008, 108, 393–399 CrossRef.
- H. I. Oyama, Polymer, 2009, 50, 747–751 CrossRef CAS PubMed.
- Z. Su, Q. Li, Y. Liu, G. H. Hu and C. Wu, Eur. Polym. J., 2009, 45, 2428–2433 CrossRef CAS PubMed.
- S. Sun, M. Zhang, H. Zhang and X. Zhang, J. Appl. Polym. Sci., 2011, 122, 2992–2999 CrossRef CAS.
- Y. P. Hao, H. H. Ge, L. J. Han, H. L. Zhang, L. S. Dong and S. L. Sun, Chin. J. Polym. Sci., 2013, 31, 1519–1527 CrossRef CAS PubMed.
- Y. Hao, H. Liang, J. Bian, S. Sun, H. Zhang and L. Dong, Polym. Int., 2014, 63, 660–666 CrossRef CAS.
- V. H. Orozco, W. Brostow, W. Chonkaew and B. L. Lopez, Macromol. Symp., 2009, 277, 69–80 CrossRef CAS.
- C. S. Yoon and D. S. Ji, Fibers Polym., 2003, 4, 59–65 CrossRef CAS.
- M. B. Coltelli, C. Toncelli, F. Ciardelli and S. Bronco, Polym. Degrad. Stab., 2011, 96, 982–990 CrossRef CAS PubMed.
- S. Lin, W. Guo, C. Chen, J. Ma and B. Wang, Mater. Des., 2012, 36, 604–608 CrossRef CAS PubMed.
- T. Sadik, F. Becquart, J. C. Majeste and M. Taha, Mater. Chem. Phys., 2013, 140, 559–569 CrossRef CAS PubMed.
- V. T. Phuong, M. B. Coltelli, P. Cinelli, M. Cifelli, S. Verstichel and A. Lazzeri, Polymer, 2014, 55, 4498–4513 CrossRef CAS PubMed.
- C. M. Thurber, Y. Xu, J. C. Myers, T. P. Lodge and C. W. Macosko, ACS Macro Lett., 2015, 4, 30–33 CrossRef CAS.
- H. Liu, F. Chen, B. Liu, G. Estep and J. Zhang, Macromolecules, 2010, 43, 6058–6066 CrossRef CAS.
- H. Liu, W. Song, F. Chen, L. Guo and J. Zhang, Macromolecules, 2011, 44, 1513–1522 CrossRef CAS.
- H. Liu, L. Guo, X. Guo and J. Zhang, Polymer, 2012, 53, 272–276 CrossRef CAS PubMed.
- Y. Chen, D. Yuan and C. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 3811–3816 CAS.
- H. Fang, F. Jiang, Q. Wu, Y. Ding and Z. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 13552–13563 CAS.
- G. C. Liu, Y. S. He, J. B. Zeng, Y. Xu and Y. Z. Wang, Polym. Chem., 2014, 5, 2530–2539 RSC.
- Y. S. He, J. B. Zeng, G. C. Liu, Q. T. Li and Y. Z. Wang, RSC Adv., 2014, 4, 12857–12866 RSC.
- G. C. Liu, Y. S. He, J. B. Zeng, Q. T. Li and Y. Z. Wang, Biomacromolecules, 2014, 15, 4260–4271 CrossRef CAS PubMed.
- W. H. Hoidy, E. A. J. Al-Mulla and K. W. Al-Janabi, J. Polym. Environ., 2010, 18, 608–616 CrossRef CAS.
- V. Ojijo, S. S. Ray and R. Sadiku, ACS Appl. Mater. Interfaces, 2012, 4, 2395–2405 CAS.
- S. Risse, L. Tighzert, F. Berzin and B. Vergnes, J. Appl. Polym. Sci., 2014, 131, 40346 CrossRef.
- W. H. Ferreira, M. M. I. B. Carmo, A. L. N. Silva and C. T. Andrade, Carbohydr. Polym., 2015, 117, 988–995 CrossRef CAS PubMed.
- D. Wu, Y. Zhang, M. Zhang and W. Yu, Biomacromolecules, 2009, 10, 417–424 CrossRef CAS PubMed.
- J. Odent, Y. Habibi, J.-M. Raquez and P. Dubois, Compos. Sci. Technol., 2013, 84, 86–91 CrossRef CAS PubMed.
- O. Monticelli, M. Calabrese, L. Gardella, A. Fina and E. Gioffredi, Eur. Polym. J., 2014, 58, 69–78 CrossRef CAS PubMed.
- A. M. El-Hadi, Polym. Eng. Sci., 2011, 51, 2191–2202 CAS.
- S. Buddhiranon, N. Kim and T. Kyu, Macromol. Chem. Phys., 2011, 212, 1379–1391 CrossRef CAS.
- S. W. Kuo, C. F. Huang, Y. C. Tung and F. C. Chang, J. Appl. Polym. Sci., 2006, 100, 1146–1161 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |