
Domino-synthesis and fluorescence properties of 4-cyano-2-oxo-1,2-dihydropyridine-3-carboxamides and 2-oxo-1,2-dihydropyridine-3,4-dicarbonitriles†
Abstract
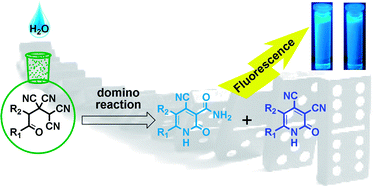
Non-catalytic conversion of 4-oxoalkane-1,1,2,2-tetracarbonitriles in the presence of water leads to the formation of a mixture of fluorescent 4-cyano-2-oxo-1,2-dihydropyridine-3-carboxamides and 2-oxo-1,2-dihydropyridine-3,4-dicarbonitriles in equal proportions. This transformation was explained, spectral-luminescence properties were investigated, and fluorescence quantum yield was measured.
 Please wait while we load your content...
Please wait while we load your content...

