
Improved oral bioavailability of novel antithrombotic S002-333 via chitosan coated liposomes: a pharmacokinetic assessment†
Abstract
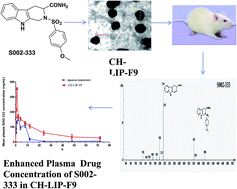
S002-333, a novel anti-thrombotic agent, exhibits excellent platelet mediated antithrombotic action and subsequently has no effect on the coagulation cascade. However, its oral bioavailability is hampered due to inherent low aqueous solubility. In order to circumvent this issue, chitosan coated liposomes were prepared by an ethanol injection method. S002-333 loaded liposomes (CH-LIP-F9) were reproduced with homogeneous particle sizes. The liposomal formulation was characterized with respect to size and surface morphology by transmission electron microscopy (TEM). The optimized formulation exhibited spherical shapes with a nano-metric size (249.64 ± 10.36 nm). The percentage entrapment efficiency (% EE) offered by the various developed formulations was found to be in the range between 72.36 ± 1.76 and 76.87 ± 2.32%. An in vitro release experiment demonstrated prolonged release of S002-333 from the optimized liposomal formulation. A cytotoxicity study represented that both blank liposomes (BLK-LIP) as well as the drug bearing liposomal formulation displayed negligible toxicity towards Caco-2 cells. The results of a pharmacokinetic study indicated that liposomal formulation significantly enhanced oral absorption of S002-333 in rats (AUC0–t; 7016.02 ± 128.96 h ng mL−1) compared to its aqueous suspension (AUC0–t; 2382.02 ± 77.17 h ng mL−1). These results together elicited that the developed liposomal formulation would improve preclinical and clinical application of S002-333.
 Please wait while we load your content...
Please wait while we load your content...

