
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePropargyl ether-functionalized poly(m-phenylene): a new precursor for the preparation of polymers with high modulus and high Tg†
Yijie
Luo
a,
Jing
Sun
*a,
Kaikai
Jin
a,
Jiajia
Wang
a,
Guangsu
Huang
b and
Qiang
Fang
*a
aKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China. E-mail: sunjing@sioc.ac.cn; qiangfang@sioc.ac.cn; Fax: +86-22-54925519; Tel: +86-21-54925337
bState Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, P. R. China
First published on 24th February 2015
Abstract
A propargyl ether-functionalized poly(m-phenylene) (PE-PMP) is reported here. This polymer exhibits good solubility and film-forming ability. After postpolymerization at high temperature, the polymer transforms to a cross-linked network, which shows high thermostability with a 5% weight loss temperature at 471 °C and a char yield of 67% at 1000 °C under N2. Thermo-mechanical analysis (TMA) reveals that the cured polymer shows an average linear coefficient of thermal expansion (CTE) of 30.6 ppm °C−1 ranging from 30 to 300 °C and a glass transition temperature (Tg) near 330 °C. Moreover, even at temperatures up to 300 °C, the cured polymer possesses a storage modulus exceeding 4.0 GPa. These data are superior to those of the commercial epoxy and novolac resins and polyimides. Furthermore, the cured polymer film has good mechanical properties with hardness, Young's modulus and a bonding strength to a silicon wafer of 1.22, 9.44 and 0.78 GPa, respectively. The cured polymer film also shows good dielectric properties with an average dielectric constant of 2.93 in a range of frequencies from 2 MHz to 30 MHz. Such results suggest that the polymer is a useful precursor for preparation of insulating materials with high modulus and Tg in the microelectronics industry.
Introduction
Much attention has been paid to polymers consisting of para-linked phenylene (PPPs) units in their main chains in the past decades due to their excellent optical and electronic properties.1–12 These polymers usually are electrochemically active, and show light emitting properties. Therefore, they have been successfully developed and widely used as the optoelectronic materials in industry.In comparison with PPPs, the polymers consisting of meta-linked phenylene units in their main chains (PMPs) have gathered an increasing interesting in the recent years.13–21 Quantum-chemical study reveals that PMPs prefer helical conformations with a helical angle of 72° between two benzene rings in the solid state,22 which endow the polymers with unique properties such as good solubility and toughness benefiting from the flexible chains. In particular, the introduction of some special side chains into the backbone of PMPs makes the polymers have interesting properties including helical folding behavior15–17 and excellent triplet emission properties.18 These good properties imply that PMPs may have the potential applications in the fields of biology15–17 and information.16,17
However, the above-mentioned PMPs usually have low molecular weight.20,23–25 The low molecular weight will generally lead to the low transition temperatures and the mechanical properties, which restricts the useful commercial applications of the polymer materials. Thus, great efforts have been made to improve the molecular weight and expand application of PMPs. For example, A. D. Schlüter and coworkers14 investigated the copolymerization of p-phenylene and m-phenylene monomers under Suzuki coupling condition. J. Sakamoto group19 developed a new route, in which an AB monomer containing both boric ester and bromo group was employed. After treated the AB monomer under the improved Suzuki coupling condition, PMPs with high molecular weight were obtained. Nevertheless, in some cases, the Suzuki coupling gave the polymers consisting of cyclic oligomers and open-chain fractions.19 Undoubtedly, further investigation on the synthesis and application of high molecular weight PMPs is still desirable.
To the best of our knowledge, a simple and efficient strategy to improve molecular weight of the polymers is postpolymerization.26–28 For instance, introducing the thermally crosslinkable groups into the backbone of PMPs could greatly amplify the molecular weight of the polymer during postpolymerization at high temperature. It is noted that propargyl ether is one of the frequently-used thermally crosslinkable groups and the formed cross-linked networks have high thermostability and good mechanical properties.29,30 The characteristic of propargyl group inspires us to introduce this group into the backbone of PMPs. We hence design and synthesize a new PMPs consisting of propargyl ether as the side chains. The new polymer exhibits good film-forming ability and solubility. After heated at high temperature, PE-PMP film transforms to a cross-linked network, which shows low coefficient of thermal expansion (CTE) ranging from 30 to 300 °C. In particular, the cured PE-PMP shows high glass transition temperature (Tg) and high modulus. These data are superior to those of the commercial epoxy and novolac resins and polyimides.31 Moreover, the cured PE-PMP possesses low dielectric constant in a range of frequencies from 2 MHz to 30 MHz.
It is noted that the high-performance polymers should possess the characteristics, including high mechanical stiffness, low dielectric constant and coefficient of thermal expansion (CTE) and easy processability.32 The good properties that the cured PE-PMP showed suggest that PE-PMP could be a useful precursor used as insulative coatings for electrical wires and electronic packaging, as well as the based matrix for preparation of advanced printed circuit boards composites.
Results and discussion
Synthesis and characterization
Scheme 1 depicts a route for the preparation of propargyl ether-functionalized PMP (PE-PMP). As shown in Scheme 1, monomer 1 was prepared in a high yield by a reaction between 4-methoxyphenol and 1,3,5-trichlorobenzene in the presence of a strong inorganic base at high temperature. The polymerization of monomer 1 was carried out by using a nickel-mediated homo-coupling reaction33 in DMAc. The obtained polymer 2 showed the number-average molecular weight (Mn) and polydispersity index (PDI) of 3500 and 1.64, respectively. Such a Mn is consistent with the result previously reported.18,23,25 The polymer with such a molecular weight has good solubility in organic solvents for the further processing. By a simple demethylation reaction, polymer 2 was transformed to polymer 3. Finally, polymer PE-PMP was successfully synthesized with a Mn and PDI of 5050 and 1.50, respectively. In comparison with the starting polymer 2, the target polymer PE-PMP showed more repeating units (23 for polymer PE-PMP and 18 for polymer 2). Such difference was attributed to the multi-step precipitation routes, in which the low molecular fractions were removed. | ||
| Scheme 1 Synthesis of PE-PMP. | ||
PE-PMP had good solubility in common organic solvents such as CHCl3, THF, toluene, DMF, acetone, hexane and cyclohexanone. Casting or spin-coating from a solution of PE-PMP in toluene gave flat and transparent films (see ESI, Fig. S1†).
The chemical structure of PE-PMP was confirmed by 1H NMR, 13C NMR, FT-IR spectra and elemental analysis (EA). The detailed data are listed in the experimental section. Fig. 1 exemplifies the 1H and 13C NMR of PE-PMP. As can be seen from Fig. 1, all data are consistent with the proposed structure.
 | ||
| Fig. 1 1H NMR (A, 500 MHz, CDCl3) and 13C NMR spectra (B, 126 MHz, CDCl3) of PE-PMP. The asterisks indicate CHCl3 and the impurity in CDCl3, respectively. | ||
Postpolymerization of PE-PMP
At high temperature, aryl propargyl ether has a tendency to form chromene through [3,3]-sigmatropic rearrangement.34 For PE-PMP, the thermal crosslinking of propargyl ether units makes the polymer convert to cross-linked network. Such a cross-linking reaction29,30,34 (or postpolymerization) is depicted in Scheme 2, and it is monitored by DSC traces (Fig. 2). As shown in Fig. 2, PE-PMP softens at 73 °C and starts the cross-linking reaction at about 212 °C. The polymer gives a maximum exothermic peak at 268 °C, similar to the curing behaviors of the polymers containing propargyl ether units.29,30 The curing degree of PE-PMP was also examined by DSC. Fig. 2 exhibits the DSC trace (red line) of the postpolymerized PE-PMP, which have been cured at 320 °C for 3 h. Obviously, the straight DSC trace implies that PE-PMP has been fully converted to the cross-linked network. In other words, the postpolymerization of PE-PMP can be achieved at 320 °C for 3 h.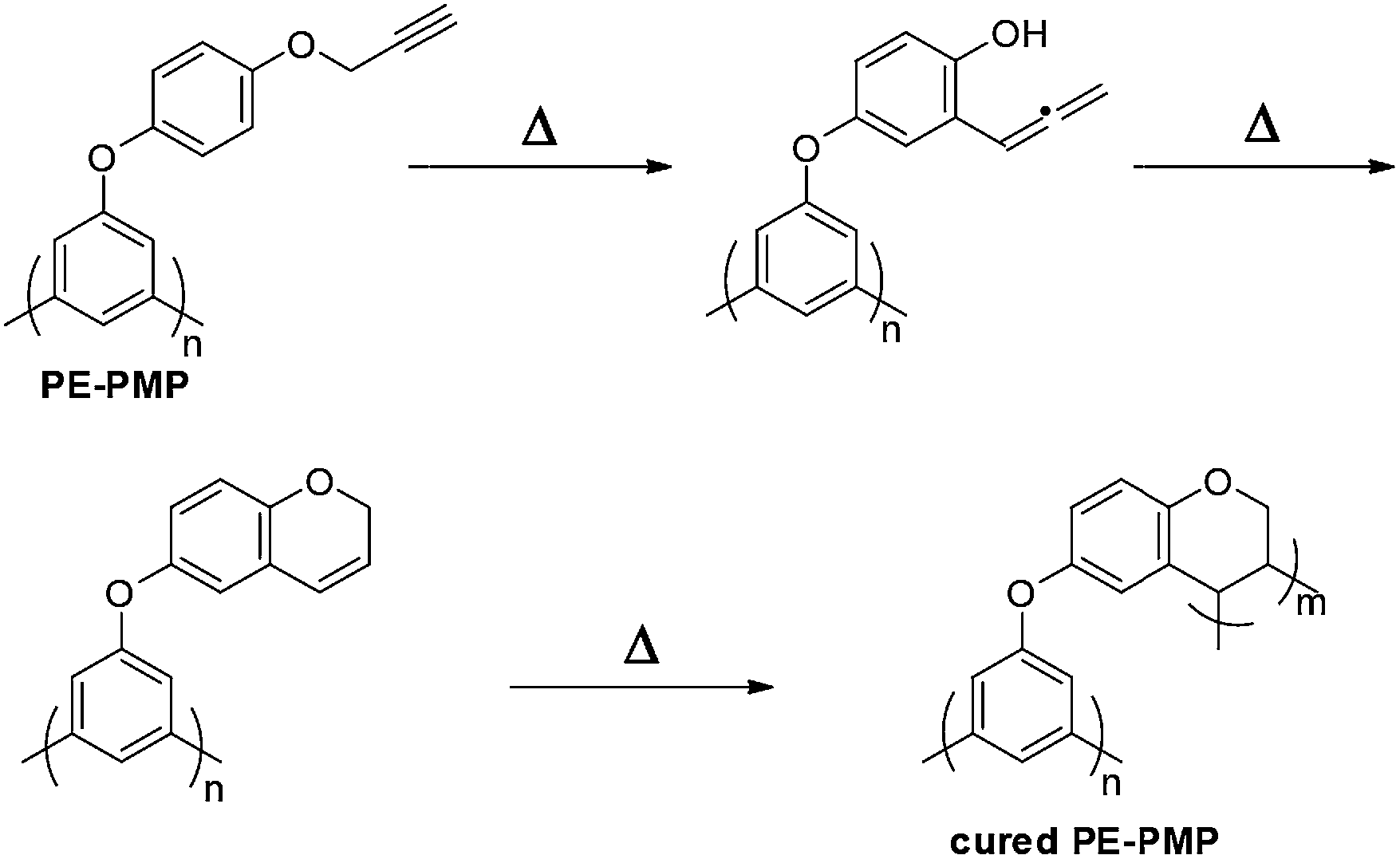 | ||
| Scheme 2 Postpolymerization reaction of PE-PMP. | ||
 | ||
| Fig. 2 DSC trace of PE-PMP at a heating rate of 10 °C min−1 in N2. | ||
The curing degree of PE-PMP was also evaluated by FT-IR spectra. Fig. 3 depicts a comparison of the FT-IR spectra between PE-PMP and cured PE-PMP. After postpolymerization, the characteristic absorption peak of alkyne group both in 3292 cm−1 (C–H stretching vibration) and 2123 cm−1 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching vibration) completely disappeared, indicating the completion of curing process.
C stretching vibration) completely disappeared, indicating the completion of curing process.
 | ||
| Fig. 3 FT-IR spectra of PE-PMP and cured PE-PMP. | ||
Thermostability of the cured PE-PMP
Thermostability of cured PE-PMP was evaluated by TGA in nitrogen and air atmosphere, and the results are depicted in Fig. 4. As shown in Fig. 4, cured PE-PMP exhibits 5 wt% loss in nitrogen and air at 471 °C and 451 °C, respectively. It is noteworthy that there is only 20 °C decreased from nitrogen to air in temperature of the 5 wt% loss, which means that the material shows high thermostability in air and could be applied in fire-resistant field. Moreover, the cured polymer shows a char yield of 67% in nitrogen at 1000 °C. These data indicate that cured PE-PMP has high thermostability. | ||
| Fig. 4 TGA curves of cured PE-PMP at a heating rate of 10 °C min−1. | ||
Mechanical properties of cured PE-PMP
High glass-transition temperature (Tg) and low coefficient of thermal expansion (CTE) are the important characteristics of high-performance engineering materials. For our newly designed material PE-PMP, the mechanical properties were measured by dynamic mechanical analysis (DMA) and thermo mechanical analysis (TMA).Fig. 5 shows the DMA results of cured PE-PMP. For comparison, two types of common commercial epoxy resins (DER331 and DEN431) were chosen, which have been widely utilized in electric and microelectronic industry. As can be seen from Fig. 5, cured PE-PMP possesses high storage modulus about 7.5 GPa at room temperature and slowly decreases to 4.0 GPa with the temperature increasing to 300 °C. Meanwhile, cured PE-PMP shows no obvious glass transition behavior even at the temperature of 300 °C. In contrast, DDS (4,4′-diaminodiphenyl sulfone) cured DER331 and DEN431 show low storage modulus (about 2.0 GPa) at room temperature and somewhat low glass transition temperature (215 °C for DER331 and 225 °C for DEN431).
 | ||
Fig. 5 DMA curves of cured PE-PMP, DER331 and DEN431. The mass ratio of DER331 (or DEN431) to DDS in curing is 3.16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. :1. | ||
It is noted that coefficient of thermal expansion (CTE) is an important factor that describes how the size of an object changes with a change of temperature. For the isotropic materials, linear CTE is to identify the ability of thermal expansion of a material. For cured PE-PMP, X-ray diffraction (XRD) (see ESI, Fig. S2†) shows that the polymer is amorphous, suggesting that such material is isotropic. Fig. 6 exhibits the relative length changes (dL/L) of a cured PE-PMP matrix (having a size of 5 mm × 3 mm × 2 mm) with the temperature. Here, the CTE values are equal to the slope according to the equation dL/L = αLdT, where αL represents linear CTE of a material and dT is the differential coefficient of the temperature, respectively. As shown in Fig. 6, cured PE-PMP shows an average linear CTE of 30.6 ppm °C−1 in a range of temperatures from 30 to 300 °C. This CTE is comparable to that of metal plumbum and better than those of the commercial epoxy resins. It is noteworthy that an interesting phenomenon occurs when the temperature rises above 150 °C, the CTE of cured PE-PMP decreases to 21.3 ppm °C−1. Moreover, cured PE-PMP shows a high Tg (near 330 °C), which is much higher than that of the commercial epoxy resins. From an application point of view, the materials with low CTE and high Tg are very desirable for their application in electrical and microelectronic industry.
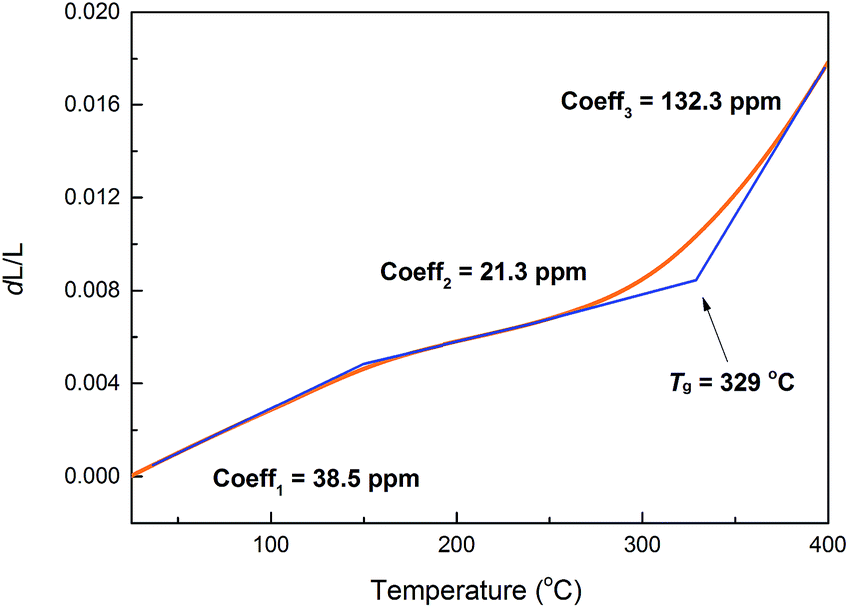 | ||
| Fig. 6 Relative length change (dL/L) versus temperature (T) of cured PE-PMP. | ||
The mechanical properties of cured PE-PMP film were also measured by nanoindentation test. The results show that the film has an average hardness of 1.22 GPa, a Young's modulus of 9.44 GPa and a bonding strength to the silicon wafer of 783 MPa, respectively. The obtained data indicated that the polymer have good mechanical properties.
Dielectric properties of cured PE-PMP
The dielectric properties of cured PE-PMP were conducted by the capacitance method.35 The device was prepared by pre-curing of PE-PMP at 150 °C in vacuum followed by postpolymerization to obtain a dark brown product. The results are illustrated in Fig. 7. As can be seen from Fig. 7, cured PE-PMP exhibits dielectric constants (k) values in a range of 2.84 to 3.02 along with dissipation factors (tanδ) below 0.014, ranging the frequencies from 2 MHz to 30 MHz. Such low k and tanδ values are also desirable in electrical and microelectronic industry.
 | ||
| Fig. 7 Dielectric constants (k) and dissipation factors (tanδ) of cured PE-PMP. | ||
Conclusions
A new poly(m-phenylene) functionalized with propargyl ether has been successfully synthesized. This polymer shows good solubility in the common organic solvents and good film-forming ability. After postpolymerization at high temperature, the polymer converts into a cross-linked network, which shows high thermostability, modulus and Tg, as well as low dielectric constant and good mechanical properties. Those data imply that the new polymer can be used as a matrix resin for the preparation of high performance composite applied in microelectronic and aerospace industry.Experimental section
Materials and instrumentation
All starting chemicals were purchased from Aldrich and used without further purification unless otherwise stated. The solvents utilized in this contribution were distilled from calcium hydride and stored under an argon atmosphere. Two commercial epoxy resins, DER331 (an bisphenol A type epoxy resin with epoxy equivalent weight of 182–192 g eq.−1) and DEN431 (an novolac type epoxy resin with epoxy equivalent weight of 172–179 g eq.−1) were purchased from The Dow Chemical Company and used as received.1H NMR and 13C NMR spectra were obtained on a Bruker AV-400 or Agilent 500/54/ASP instrument. FT-IR spectra were recorded on a Thermo Scientific Nicolet spectrometer with KBr pellets. Differential Scanning Calorimetry (DSC) was determined with TA Instrument DSC Q200 at a heating rate of 10 °C min−1 under nitrogen flow. Thermogravimetric analysis (TGA) was performed on a TG 209F1 apparatus with a heating rate of 10 °C min−1 in nitrogen or air atmosphere. Molecular weights were measured by using a Waters Breeze2a 200 GPC instrument with polystyrene as standards and tetrahydrofuran (THF) as the eluent at 35 °C. The dielectric constants (k) of the cured samples were measured in the range of frequencies from 2 MHz to 30 MHz at room temperature by a 4294A Precision Impedance Analyzer (Agilent). The surface toughness of the cured polymer film was measured by atom force microscopy (AFM) (Shimadzu SPM-9500J3). A TI 950 TriboIndenter (Hysitron) was used to measure the mechanical properties of films including hardness, Young's modulus and bonding strength. Dynamic mechanical analysis (DMA) and thermo-mechanical analysis (TMA) were recorded on the Mettler Toledo DMA/SDTA861e and TMA/SDTS841e instruments, respectively with a heating rate of 3 °C min−1 in air.
Synthesis
Acknowledgements
Financial supports from the Natural Science Foundation of China (NSFC, no. 21374131), the Shanghai Natural Science Foundation (no. 15ZR1449200) and the Opening Project of State key laboratory of Polymer Materials Engineering (Sichuan University, Grant no. sklpme2014-4-22) are gratefully acknowledged.References
- P. Kovacic and M. B. Jones, Chem. Rev., 1987, 87, 357–379 CrossRef CAS.
- J. M. Tour, Adv. Mater., 1994, 6, 190–198 CrossRef CAS.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- L.-H. Xie, C.-R. Yin, W.-Y. Lai, Q.-L. Fan and W. Huang, Prog. Polym. Sci., 2012, 37, 1192–1264 CrossRef CAS.
- L. Chen, Y. Chen, K. Yao, W. Zhou, F. Li, L. Chen, R. Hu and B. Z. Tang, Macromolecules, 2009, 42, 5053–5061 CrossRef CAS.
- P. Baum, W. H. Meyer and G. Wegner, Polymer, 2000, 41, 965–973 CrossRef CAS.
- Y. Takeoka, K. Umezawa, T. Oshima, M. Yoshida, M. Yoshizawa-Fujita and M. Rikukawa, Polym. Chem., 2014, 5, 4132–4140 RSC.
- M. R. Talipov, A. Boddeda, Q. K. Timerghazin and R. Rathore, J. Phys. Chem. C, 2014, 118, 21400–21408 CAS.
- L. Tao, J. Ju, F. Niu, T. Wang and Q. Wang, Polymer, 2013, 54, 5358–5362 CrossRef CAS.
- C. Schwarz, H. Bässler, I. Bauer, J. M. Koenen, E. Preis, U. Scherf and A. Köhler, Adv. Mater., 2012, 24, 922–925 CrossRef CAS PubMed.
- S. Schmücker and D. Kuckling, Macromol. Chem. Phys., 2012, 213, 1725–1734 CrossRef.
- Y. Y. Durmaz, E. L. Sahkulubey, Y. Yagci, E. Martinelli and G. Galli, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4911–4919 CrossRef CAS.
- P. Rattanatraicharoen, Y. Tanaka, K. Shintaku, T. Kawaguchi, K. Yamabuki, T. Oishi and K. Onimura, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1315–1322 CrossRef CAS.
- R. Kandre, K. Feldman, H. E. Meijer, P. Smith and A. D. Schlüter, Angew. Chem., Int. Ed., 2007, 46, 4956–4959 CrossRef CAS PubMed.
- H. Katagiri, T. Miyagawa, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2006, 45, 1741–1744 CrossRef CAS PubMed.
- T. Ben, H. Goto, K. Miwa, H. Goto, K. Morino, Y. Furusho and E. Yashima, Macromolecules, 2008, 41, 4506–4509 CrossRef CAS.
- T. Ben, Y. Furusho, H. Goto, K. Miwa and E. Yashima, Org. Biomol. Chem., 2009, 7, 2509–2512 CAS.
- J. Liu and Q. Pei, Macromolecules, 2010, 43, 9608–9612 CrossRef CAS.
- B. Hohl, L. Bertschi, X. Zhang, A. D. Schlüter and J. Sakamoto, Macromolecules, 2012, 45, 5418–5426 CrossRef CAS.
- J. Tong, S. Diao, K. Jin, C. Yuan, J. Wang, J. Sun and Q. Fang, Polymer, 2014, 55, 3628–3633 CrossRef CAS.
- X. Zhang, L. Sheng, T. Higashihara and M. Ueda, Polym. Chem., 2013, 4, 1235–1242 RSC.
- S. Y. Hong, D. Y. Kim, C. Y. Kim and R. Hoffmann, Macromolecules, 2001, 34, 6474–6481 CrossRef CAS.
- L. J. Mathias and G. L. Tullos, Polymer, 1996, 37, 3771–3774 CrossRef CAS.
- J. L. Reddinger and J. R. Reynolds, Macromolecules, 1997, 30, 479–481 CrossRef CAS.
- T. Yamamoto, M. Abe, B. Wu, B. K. Choi, Y. Harada, Y. Takahashi, K. Kawata, S. Sasaki and K. Kubota, Macromolecules, 2007, 40, 5504–5512 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H. A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- J. Romulus, J. T. Henssler and M. Weck, Macromolecules, 2014, 47, 5437–5449 CrossRef CAS.
- C. Yuan, J. Wang, K. Jin, S. Diao, J. Sun, J. Tong and Q. Fang, Macromolecules, 2014, 47, 6311–6315 CrossRef CAS.
- T. Agag and T. Takeichi, Macromolecules, 2001, 34, 7257–7263 CrossRef CAS.
- K. D. Demir, B. Kiskan and Y. Yagci, Macromolecules, 2011, 44, 1801–1807 CrossRef.
- D.-J. Liaw, K.-L. Wang, Y.-C. Huang, K.-R. Lee, J.-Y. Lai and C.-S. Ha, Prog. Polym. Sci., 2012, 37, 907–974 CrossRef CAS.
- W. R. Shiang and E. P. Woo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2081–2091 CrossRef CAS.
- V. Percec, J. Y. Bae, M. Zhao and D. H. Hil, Macromolecules, 1995, 28, 6726–6734 CrossRef CAS.
- J. Zsindely and H. Schmidt, Helv. Chim. Acta, 1968, 51, 1510–1514 CrossRef CAS.
- C. Yuan, K. Jin, K. Li, S. Diao, J. Tong and Q. Fang, Adv. Mater., 2013, 25, 4875–4878 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01515d |
| This journal is © The Royal Society of Chemistry 2015 |