DOI:
10.1039/C5RA01466B
(Paper)
RSC Adv., 2015,
5, 50353-50364
A DFT/TDDFT mission to probe push–pull vinyl coupled thiophene oligomers for optoelectronic applications†
Received
25th January 2015
, Accepted 15th May 2015
First published on 21st May 2015
Abstract
Vinyl coupled thiophene oligomers (VCTOs) are one of the active components in organic solar cells. In the present study, VCTOs with various acceptor groups (–CN, –NO2 and –COOH) have been considered and their optoelectronic properties have been evaluated using DFT/TDDFT calculations. In total, 17 VCTOs, including 3 already reported, have been considered. The computed results reveal that the reference VCTOs (VCTO1, 2 and 3) can be used as possible electron transport materials, and the newly designed VCTOs are found to be promising hole transport materials. Among these, VCTO4b is found to show a lower band gap, whereas VCTO3c has a higher band gap. Furthermore, the study explores the roles of donor and acceptor groups in the band gap, ionization potential, electron affinity, exciton binding energy and light harvesting efficiency of these VCTOs. The spectral analysis shows that the modelled VCTOs have strong n → π* transitions, while the reference VCTOs were found to show predominant π → π* transitions. In summary, 9 out of 14 of the designed VCTOs were found to show better optoelectronic properties than their reference molecules.
1. Introduction
The design and development of renewable energy sources using organic solar cells (OSC) have attracted considerable interest in recent years due to increasing energy demand.1–7 Owing to their large surface areas and porous structures, metal organic frameworks (MOF) and covalent organic frameworks (COF) have been the subject of research in the areas of gas storage and catalysis.8–12 Quantum chemical calculations have been used to understand not only the nature of MOFs/COFs (both in the ground and excited states) but also the character of their bands.13–20 Their unique blend of optical and electronic properties has led to their association with the areas of photocatalysis, photovoltaics and electrochemical devices.21,22 OSC research is currently undergoing rapid growth due to the cost effective production of OSCs by vacuum processing on flexible substrates and their higher power conversion efficiencies (PCE) of 10.7% in an active area of 1.1 cm2.23 Typically, solution processed solar cells with a nano phase separated blend of a semiconducting polymer as the donor and soluble fullerene as the acceptor afford PCE values exceeding 8%.24 In this context, OSCs made from π-conjugated oligomers or dyes appear to be one of the most promising materials in the conversion of solar energy into electricity. The intensive research effort to prepare conjugated polyheterocycles by electropolymerisation originated in 1979.25 Polypyrrole was prepared using pyrrole by an electropolymerisation method.25,26 Later, this was extended to thiophene,27–29 furan,30 indole,31 carbazole,32 benzene33 and fluorene.34 Among these polyheterocycles, thiophene oligomer is a subject of considerable interest due to its simple structure, and these oligomers have been used as models35–40 to study the electronic properties of polythiophene.41–47 Therefore, the use of thiophene based molecules is inevitable in the fabrication of materials for solar cells.48 Recent developments in small molecule organic solar cells (SMOSC) mainly use these conjugated oligomers.49
Recently, a series of π-conjugated oligomers were synthesized by Peter Bauerle et al.23 and were reported to have a high PCE of 6.9%. This group has synthesized a novel series of π-conjugated vinyl coupled thiophene oligomers (VCTO), consisting of electron withdrawing molecules containing –CN groups on either side of the molecule, for photovoltaic (PV) applications.23 It is well known that thiophene oligomers have excellent conjugation, planarity and rigidity in their rings but have structures that can be flexibly tuned for better photovoltaic performance. This prompted us to substitute various electron releasing and withdrawing groups on VCTOs and screen the resulting compounds for their optoelectronic properties. Particularly, the effects of electron releasing groups, electron withdrawing groups and π-bridges on the optoelectronic properties of the VCTOs was analyzed using modern computational tools. A set of VCTOs have been designed with electron releasing groups and electron withdrawing groups at their terminal ends, and their suitability for optoelectronic applications has been examined. Their properties have been computed and compared with available experimental results.
In this work, the structure and properties of VCTOs have been systematically analyzed via quantum chemical calculations using density functional theory (DFT) and time dependent density functional theory (TDDFT) methods. Their optimized geometries, electronic properties, frontier molecular orbital analyses (FMO), energy gaps, ionization energies (IP), electroaffinities (EA), light harvesting efficiencies (LHE), excitation binding energies (Eb), hyperpolarizabilities (β°) and charges have been calculated and analyzed. NBO analysis has helped us to ascertain the most stable donor–acceptor interactions in these molecules, and the effect of solvation has been studied using Polarizable Continuum Model (PCM) calculations. The results for each compound are discussed here.
2. Computational details
The structures of the various thiophene oligomers chosen for this work are shown in Fig. 1. The B3LYP functional50,51 is known to perform well for most organic molecules, and therefore the same is adopted here.52–54 The ground state geometries of the molecules have been optimized using a hybrid exchange-correlation B3LYP functional with the 6-31g (d) basis set. All stationary points have been confirmed as minima in the potential energy surface (PES) by frequency analysis. The computed and experimental values of bond lengths and angles are in agreement, which emphasizes the reliability of the DFT method [SIT1]. The first order hyperpolarizability has been computed to determine the NLO response of each molecule. The first order hyperpolarizability (β0) has been calculated at the 6-31g (d) level. From this βtotal, a scalar quantity can be computed, as reported earlier,27,54,55 from the X, Y and Z components of β using eqn (1):| | |
βtot = (βx2 + βy2 + βz2)1/2
| (1) |
where, βx2= (βxxx + βxyy + βxzz)2, βy2= (βyyy + βyzz + βyxx)2 and βz2= (βzzz + βzxx + βzyy)2.
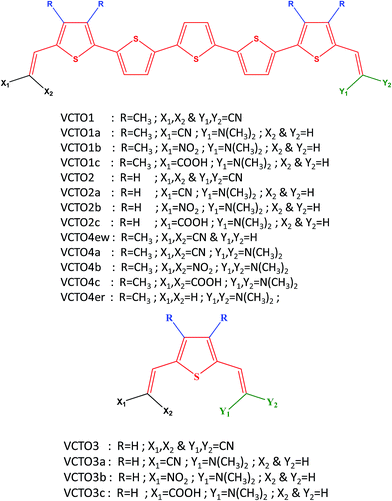 |
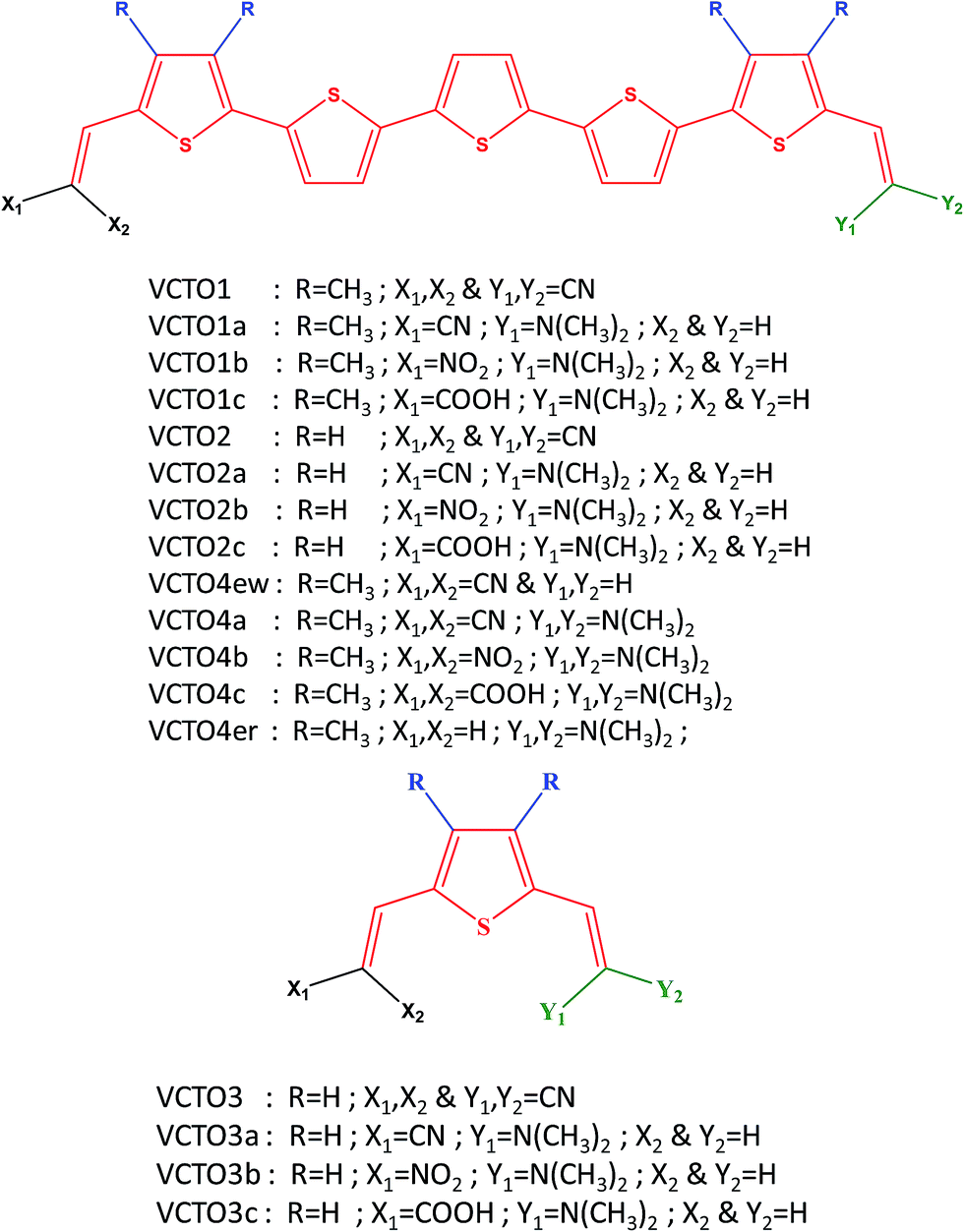
| | Fig. 1 Structure of the designed and reference VCTO molecules. | |
Time dependent density functional theory (TDDFT) has been employed here to calculate the excitation energies, absorption wavelengths, and oscillator strengths of the ground state optimized geometries at the 6-31g (d) level.56 Peter Bauerle et al. have reported the absorption spectra in dichloromethane (DCM) medium for VCTO1, 2, and 3, and TDDFT calculations have been performed at the M062X level in the same medium. The Polarizable Continuum Model (PCM) has been used to find the excitation energies in the solvent medium.57 The ionization potentials (IP) and electron affinities (EA) have been calculated according to Koopman's theorem.58 Further NBO analysis59–61 has been performed at the B3LYP/6-31g (d) level to find the most stabilization ground state interactions in these molecules. All calculations have been carried out using the Gaussian 03 and 09 suites of programs.62
3. Results and discussion
In the present study, a total of 17 asymmetric VCTOs, including three (VCTO1, VCTO2 & VCTO3) that have been experimentally reported, have been considered (Fig. 1); the three reported VCTOs are taken as reference for validation. The chosen candidates fall into four categories. The first (VCTO1) is composed of 5 alternate thiophene rings with methyl groups on the 1st and 5th rings. The second category (VCTO2) of oligomer has no substitution on the 5 thiophene rings, and these can be used to determine the effects of methyl substitution. The third category (VCTO3) has only 1 thiophene ring; these can be used to analyze the effects of π conjugation with increasing number of thiophene rings. VCTO4 type candidates have been constructed using –N(CH3)2 as the donor group. VCTO4ew (electron withdrawing group) and VCTO4er (electron releasing group) were specially designed to determine the contribution of electron withdrawing groups and electron releasing groups towards the optoelectronic properties of VCTOs. This may offer valuable clues for the design of novel, more efficient molecules. All the three parent molecules (VCTO1, VCTO2 & VCTO3) have four –CN groups (Fig. 1) attached on either side of their vinyl moiety. The diverse structural features of these VCTOs prompted us to study how various substitutions alter their photo-physical properties. Therefore, fourteen new VCTO derivatives have been designed with various substitutions, as shown in Fig. 1, where a represents –CN substitution and b & c denote –NO2 and –COOH substitutions respectively. The parent molecules do not have electron push–pull character, as both sides contain –CN groups; it would be interesting to substitute an electron donor group at one end of the molecule and examine its push–pull character. In the literature, dimethyl amine54 has been found to act as a good donor group; therefore, it was used in the design of the new molecules [Fig. 1].
3.1 Ground state geometries
The optimized ground state geometries of the VCTOs are given in SIF1 (ESI Fig. 1†). The important geometrical parameters are listed in Table SIT1.† The computed and experimental bond parameters are in good agreement. For instance, the C–S bond length in VCTO3 is 1.745 Å and the calculated value is 1.744 Å; the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond length is 1.366 Å, whereas the calculated CC bond length is 1.370 Å. These results emphasize the reliability of the choice of B3LYP for this work. To understand the delocalization of the π-electrons, computed bond parameters are presented in Fig. 2. From Fig. 2, it can be observed that C–C, C–S, and C–N are all well within their single and double bond limits, respectively. This indicates the presence of extended π conjugation in these VCTOs. The C–C–C angles between each thiophene ring vary from 124–131°. The twist angle for all VCTOs lies in the range of 153–179°. After validating the DFT method, the effects of electron withdrawing and releasing groups on the bond parameters were analysed. For example, in VCTO4ew, where there is no electron releasing group, the vinylic C32C39 bond length is 1.34 Å; meanwhile, the vinylic C10C11 bond near the electron withdrawing group experiences an elongation to 1.37 Å. Moreover, in VCTO4er, the same vinylic carbon near the electron releasing group displayed an extended bond length of 1.37 Å and the C10C11 bond near the pull group showed a bond length of 1.34 Å. This shows that when the vinylic carbon is substituted by electron withdrawing and electron releasing groups, the bond length is stretched considerably, which results in single and double bond character and causes extended conjugation in the VCTOs.
C bond length is 1.366 Å, whereas the calculated CC bond length is 1.370 Å. These results emphasize the reliability of the choice of B3LYP for this work. To understand the delocalization of the π-electrons, computed bond parameters are presented in Fig. 2. From Fig. 2, it can be observed that C–C, C–S, and C–N are all well within their single and double bond limits, respectively. This indicates the presence of extended π conjugation in these VCTOs. The C–C–C angles between each thiophene ring vary from 124–131°. The twist angle for all VCTOs lies in the range of 153–179°. After validating the DFT method, the effects of electron withdrawing and releasing groups on the bond parameters were analysed. For example, in VCTO4ew, where there is no electron releasing group, the vinylic C32C39 bond length is 1.34 Å; meanwhile, the vinylic C10C11 bond near the electron withdrawing group experiences an elongation to 1.37 Å. Moreover, in VCTO4er, the same vinylic carbon near the electron releasing group displayed an extended bond length of 1.37 Å and the C10C11 bond near the pull group showed a bond length of 1.34 Å. This shows that when the vinylic carbon is substituted by electron withdrawing and electron releasing groups, the bond length is stretched considerably, which results in single and double bond character and causes extended conjugation in the VCTOs.
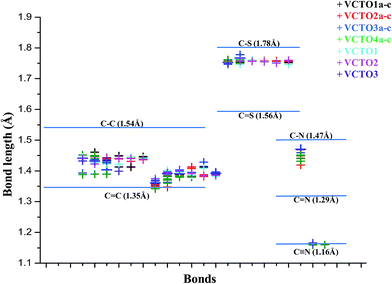 |
| | Fig. 2 Depiction of electron delocalization in VCTOs from the calculated C–C, C–N, C–S bond lengths. | |
3.2 Frontier molecular orbital analysis
The chemical reactivity and kinetic stability of a molecule can be characterized using Frontier Molecular Orbital (FMO) analysis.54,63,64 The Highest Occupied Molecular Orbital (HOMO), Lowest Unoccupied Molecular Orbital (LUMO) and HOMO–LUMO energy gap give qualitative information about the charge transfer interaction that occurs within a molecule.27,54,63,64 In order to predict the optical and electronic properties of these VCTOs, the HOMO/LUMO levels were calculated and are listed in the supporting information (SIT2) and shown in Fig. 3. It is well known that electron and hole transport materials should have suitable HOMO/LUMO levels in order to transport electrons and holes from the respective electrodes into the emitting layer of an OLED, thereby controlling the HOMO and LUMO levels; this results in molecules with desirable charge transport properties.
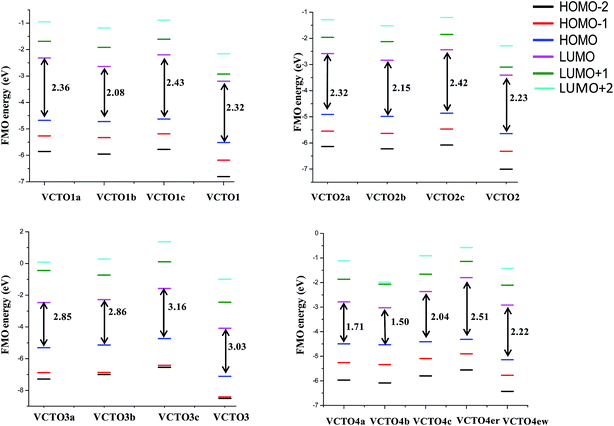 |
| | Fig. 3 Frontier molecular orbital energy level diagram of the designed VCTOs. | |
HOMOs were observed in the range of −7.11 eV to −4.31 eV and LUMOs in the range of −4.08 eV to −1.57 eV (Fig. 3). The calculated band gaps for the reported VCTOs (VCTO1, 2 & 3) are 2.32 eV, 2.23 eV and 3.03 eV respectively. It is important to note that the band gap of VCTO1 (R = –CH3) is 0.09 eV greater than that of VCTO2 (R = H). This shows that methyl substitution makes a minimal contribution to the FMOs. Out of the 14 designed VCTOs, six VCTOs were found to have band gaps lower than that of VCTO2 (2.23 eV). From Fig. 3, it is clear that VCTO4b has the lowest band gap (1.50 eV), whereas VCTO3c shows the largest band gap (3.16 eV). In the VCTO1 group, VCTO1b was found to have a band gap 0.24 eV lower than that of VCTO1. It is interesting to see that though VCTO1c has a higher HOMO (−4.63 eV), it still shows the largest band gap due to its higher LUMO (2.20 eV). Thus, the increasing order of band gaps in this VCTO1 group can be listed as VCTO1b < VCTO1 < VCTO1a < VCTO1c. The HOMO of the VCTO2 group of molecules lies over a range of −5.64 eV to −4.86 eV. The maximum HOMO–LOMO energy gap was found for VCTO2c (2.42 eV) and the minimum HOMO–LOMO energy gap was found for VCTO2b (2.15 eV). Four out of five molecules (VCTO4a, 4b, 4c & 4ew) in the VCTO4 group were found to show reduced band gaps relative to that of VCTO2. VCTO4er was found to have a higher band gap (2.51 eV) than the other molecules in the VCTO4 group. This shows that the acceptor group is very important for band gap reduction in these molecules. VCTO4b and VCTO1b differ by only one donor and acceptor group (–NO2 & –N(CH3)2), and the additional donor and acceptor group in VCTO4b play major roles in reducing the HOMO–LUMO energy gap from 2.08 eV to 1.50 eV. When comparing VCTO1a and VCTO4a, the HOMO increases to −4.49 eV from −4.67 eV while the LUMO decreases to −2.78 eV from 2.31 eV. It is evident that the additional donor and acceptor groups play vital roles in altering the FMOs. By looking at the structure of VCTO4s (Fig. 1), this group can be classified into three categories: VCTO4er (donor alone), VCTOew (acceptor alone) and VCTO4a, 4b & 4c (Donor–π–Acceptor). The increasing band gap order in the VCTO4 group is as follows: VCTO4b < VCTO4a < VCTO4c < VCTOew < VCTO4er. This shows that Donor–π–Acceptor molecules are superior to Acceptor–π–Acceptor (VCTO1, VCTO2 & VCTO3) molecules.
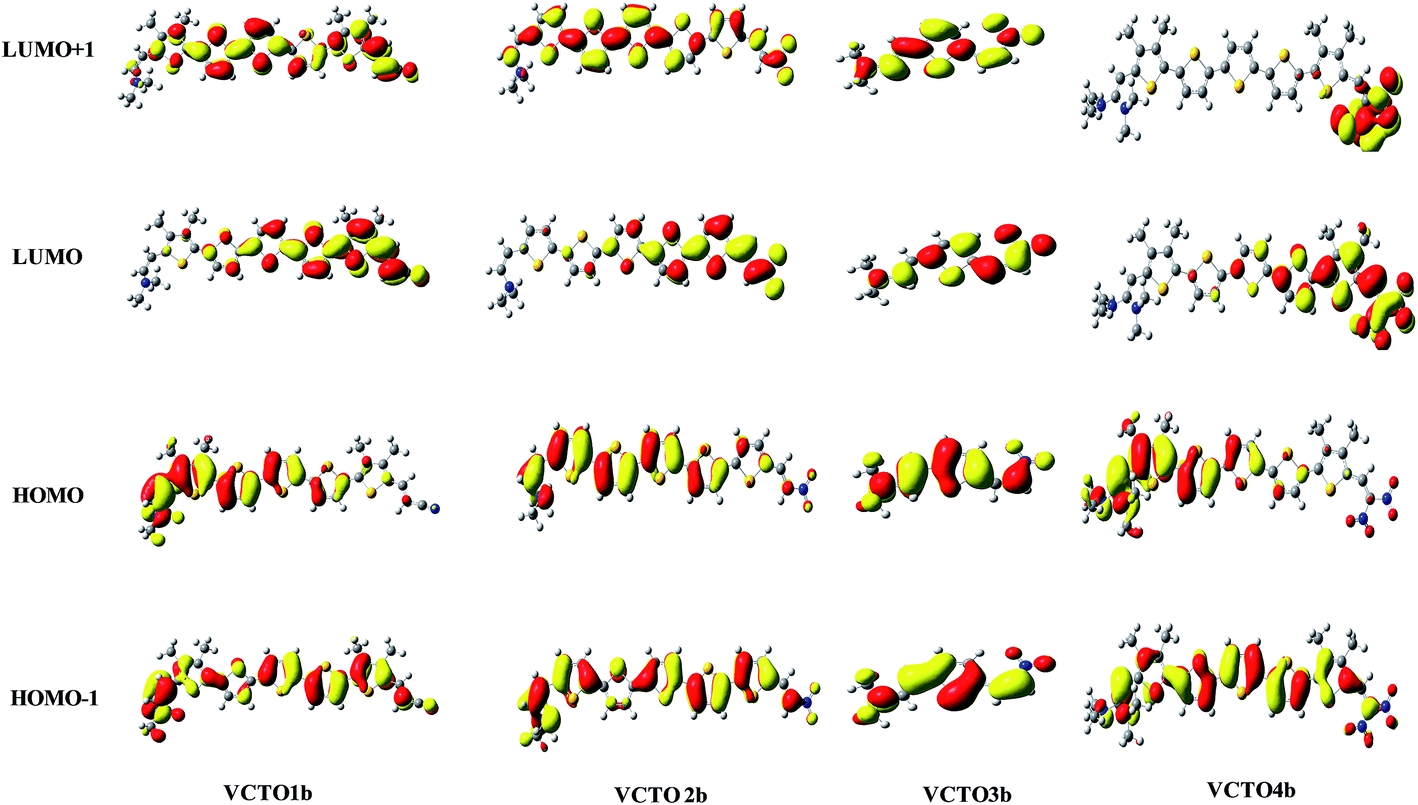
The Frontier molecular orbitals (LUMO + 1, LUMO, HOMO & HOMO − 1) of all the 'b' molecules of VCTOs (VCTO1b, 2b, 3b & 4b) are given in Fig. 4. The complete FMOs of all the VCTOs are deposited in SIF2–SIF5. From this figure, it is clear that the HOMO and LUMO of VCTO3b are evenly localized on the entire molecule. This ensures that the HOMO → LUMO transition in VCTO3b is due to a π → π* transition. In contrast to the HOMO of VCTO1b, the HOMOs of 2b & 3b are mainly localized by the donor and π-bridge units, whereas the LUMOs are largely stabilized by the acceptor groups and a few thiophene units. This indicates that the HOMO → LUMO transition arises from intra-molecular charge transfer character (IMCT). Meanwhile, the experimentally reported VCTOs (VCTO1, 2 & 3) exhibit a typical π–π* transition (SIF2–SIF4). This shows that the introduction of electron releasing groups on VCTOs causes the HOMO → LUMO transition to arise from IMCT. This IMCT character of the HOMO → LUMO transition is reflected in the absorption maxima (λmax), which is discussed in Section 3.7.
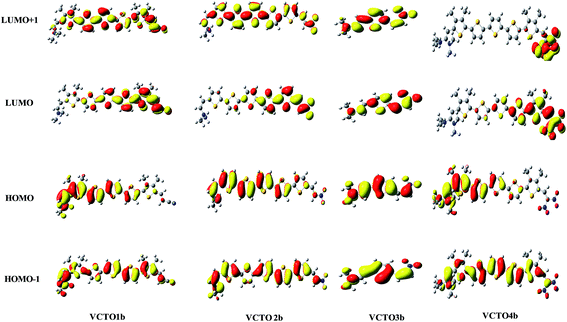 |
| | Fig. 4 Frontier molecular orbitals of the nitro derivatives of VCTOs. | |
To gain further insights from these FMOs, the energetics of the HOMOs and LUMOs were analyzed using QMForge,27,54,64 and the results are summarized in Table 1. The whole molecule is segmented into four fragments, namely the thiophene rings (π-bridge), N,N-dimethylamine (donor), –CN, –NO2, and –COOH (acceptors) and the methyl groups on the thiophene rings (methyl); their corresponding compositions have been computed. From the table, it is clear that both the HOMO and LUMO of VCTO1 receive contributions of 87% & 79% from the π-bridge units, respectively. Only 1% of the contribution towards the HOMO and LUMO of VCTO1 comes from the methyl groups, and this is also reflected in the FMOs (SIF2). The π-bridge contributes nearly ∼50% towards the HOMO and LUMO in the VCTO3s; this is due to the lesser number of thiophene units in this group. The contribution of the donor in the VCTO3s is the highest (∼35%) among the VCTOs. The lowest band gap, that of VCTO4b, receives a contribution of 27% from the donor towards the HOMO; its contribution is reduced to less than 1% in the LUMO. However, the contribution of the acceptor unit increases from 1% to 34% moving from the HOMO to the LUMO in VCTO4b. As expected, the donor contributes 24% towards the HOMO in VCTO4er, while the acceptor contributes 21% towards the stabilization of the LUMO in VCTOew. Invariably, in all the molecules, the π-bridge predominantly contributes to the HOMO & LUMO (43% to 93%). Except for the VCTO3s, the contribution of the π-bridge is at least two times higher than that of the corresponding donor and acceptor. On the whole, this study implies that the π-bridge must be substituted by electron releasing groups to alter the HOMO, while the LUMO can be altered by substituting electron withdrawing groups. This fragment analysis sheds light on how to alter HOMO and LUMO contributions by making suitable substitutions on the π bridge.
Table 1 Frontier molecular orbital composition (%) of various fragments of the VCTOs optimized at the B3LYP/6-31g(d) level
| Molecules |
Orbitals |
π bridge |
Donor |
Acceptor |
Methyl |
| VCTO1 |
HOMO |
86.94 |
0 |
11.51 |
1.55 |
| LUMO |
79.38 |
0 |
19.61 |
1.01 |
| VCTO1a |
HOMO |
75.91 |
20.88 |
0.88 |
2.33 |
| LUMO |
83.13 |
0.45 |
15.34 |
1.08 |
| VCTO1b |
HOMO |
75.26 |
21.47 |
0.90 |
2.37 |
| LUMO |
66.11 |
0.20 |
32.67 |
1.03 |
| VCTO1c |
HOMO |
77.51 |
19.10 |
1.06 |
2.33 |
| LUMO |
83.91 |
0.54 |
14.44 |
1.11 |
| VCTO2 |
HOMO |
87.62 |
0 |
12.38 |
— |
| LUMO |
80.94 |
0 |
19.06 |
— |
| VCTO2a |
HOMO |
86.54 |
11.18 |
2.28 |
— |
| LUMO |
85.44 |
1.11 |
13.45 |
— |
| VCTO2b |
HOMO |
84.88 |
13.12 |
1.99 |
— |
| LUMO |
70.85 |
0.46 |
28.67 |
— |
| VCTO2c |
HOMO |
86.66 |
10.96 |
2.36 |
— |
| LUMO |
85.38 |
1.26 |
13.28 |
— |
| VCTO3 |
HOMO |
43.16 |
0 |
56.84 |
— |
| LUMO |
55.40 |
0 |
44.60 |
— |
| VCTO3a |
HOMO |
46.71 |
32.32 |
20.97 |
— |
| LUMO |
56.76 |
18.85 |
24.40 |
— |
| VCTO3b |
HOMO |
48.88 |
35.24 |
15.88 |
— |
| LUMO |
44.35 |
14.02 |
41.63 |
— |
| VCTO3c |
HOMO |
49.22 |
35.94 |
14.84 |
— |
| LUMO |
53.55 |
18.11 |
28.34 |
— |
| VCTO4ew |
HOMO |
89.13 |
5.22 |
3.61 |
2.04 |
| LUMO |
77.63 |
0.21 |
21.00 |
1.16 |
| VCTO4a |
HOMO |
70.35 |
26.38 |
0.79 |
2.48 |
| LUMO |
77.05 |
0.39 |
21.37 |
1.19 |
| VCTO4b |
HOMO |
69.49 |
27.17 |
0.85 |
2.49 |
| LUMO |
64.96 |
0.29 |
33.65 |
1.09 |
| VCTO4c |
HOMO |
71.69 |
25.12 |
0.67 |
2.52 |
| LUMO |
77.14 |
0.42 |
21.12 |
1.32 |
| VCTO4er |
HOMO |
73.22 |
23.74 |
0.53 |
2.52 |
| LUMO |
93.40 |
1.98 |
3.68 |
0.94 |
3.3 Dipole moment
Dipole moment calculations give a clear picture of the electronic charge distribution in a molecule. Therefore, we decided to analyse and compare the dipole moments (μ) in the gas phase and solution phase (dichloromethane medium, DCM) of the VCTOs considered here (Table 2). The highest dipole moment (μ in Debye) in the gas phase is observed for VCTO4b (16.94 D). VCTO3 and VCTO3c, which have wide band gaps, have lower dipole moments (4.41 D). Table 2 shows that VCTOew and VCTOer show large (6.63 D) differences in their dipole moments. This shows that the electron withdrawing group enhances the dipole moment of the VCTOs. VCTO2 has a dipole moment of 9.37 D, which is twice that of VCTO3 (4.41 D). This is due to the presence of long π-bridge (thiophene units) in VCTO2. It is noteworthy that solvation increases the dipole moment to 7.11 D (Table 2). Solvation has the least effect (0.8 D) on the dipole moment of VCTO2c, whereas a difference of 7.11 D is observed for VCTO3 when moving from the gas to the solution phase. In general, the dipole moments of the reference molecules (VCTO1, 2 & 3) are largely affected by solvation. However, the dipole moments of all the carboxylic acid derivatives (VCTO1c, 2c, 3c & 4c) were found to show less of a response (∼1 D) to solvation. Among the designed VCTOs, the highest dipole moment of 21.20 D is exhibited by VCTO4b, whereas only 5.39 D is observed for VCTO3c. In general, the calculated dipole moments are in harmony with the calculated HOMO–LUMO energy gap.
Table 2 Dipole moments in the gas phase and in DCM medium and hyperpolarizability values at the B3LYP/6-31g(d) level
| Molecules |
Dipole moment (Debye) |
Hyperpolarizability (esu) |
| Gas phase |
CH2Cl2 |
| VCTO 1 |
12.16 |
16.13 |
50.93 |
| VCTO1a |
10.35 |
12.21 |
676.3 |
| VCTO1b |
11.47 |
13.73 |
1083 |
| VCTO1c |
6.61 |
7.59 |
595.05 |
| VCTO2 |
9.37 |
13.23 |
44.8 |
| VCTO2a |
9.65 |
11.32 |
754.69 |
| VCTO2b |
11.19 |
13.42 |
963.55 |
| VCTO2c |
6.03 |
6.83 |
499.05 |
| VCTO3 |
4.41 |
11.52 |
58.36 |
| VCTO3a |
12 |
15.95 |
55.5 |
| VCTO3b |
10.70 |
14.19 |
76.19 |
| VCTO3c |
4.41 |
5.39 |
43.69 |
| VCTO4ew |
11.44 |
14.38 |
740.90 |
| VCTO4a |
15.43 |
18.70 |
2280.90 |
| VCTO4b |
16.94 |
21.20 |
3278.83 |
| VCTO4c |
9.32 |
10.92 |
1242.28 |
| VCTO4er |
4.81 |
5.63 |
411.48 |
3.4 NLO properties
The NLO response of a molecule originates from the individual contributions of basic molecular units in an organic material. NLO activity studies emphasized a third rank tensor called first-order hyperpolarizability (β0). This can be described by a 3 × 3 × 3 matrix. This 3D matrix with 27 components can be reduced to 10 components due to Kleinman symmetry.65 We aimed to analyse and compare the electronic effects on the β0 values of VCTOs, and the results are summarized in Table 2.
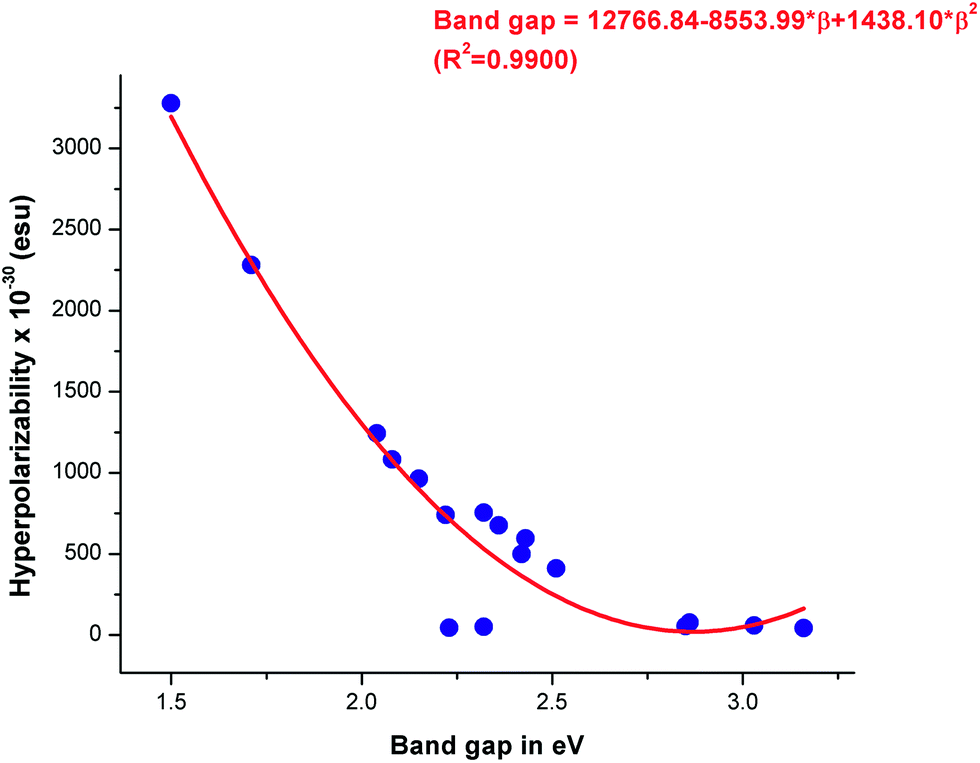
The calculated β0 values of the VCTO4s follows an increasing order of VCTO4er < VCTO4ew < VCTO4c < VCTO4a < VCTO4b. It is important to note that this is in excellent agreement with the computed NLO responses and the computed band gaps. For example, VCTO4b, which has the lowest band gap, was found to show high hyperpolarizability (3278.83 esu), whereas VCTO3c, which has the widest band gap, has a very low hyperpolarizability of 43.69 esu. It is evident from Table 2 that the first order hyperpolarizabilities of VCTO1 & 2 are much lower than those of their designed candidates. For instance, the first order hyperpolarizability of VCTO2 is 44.8 esu, and 963.55 esu is observed for VCTO2b. This shows that D–π–A type molecules are superior to A–π–A type molecules. It is interesting to note that the VCTO3s have very low hyperpolarizability values (up to ∼19 esu) even after the introduction of electron donors or acceptors. This suggests that π-conjugation is important in these systems for enhanced NLO properties. The relationship between the band gap and hyperpolarizability (β0) has been evaluated using a correlation coefficient parameter (R2) by a quadratic fitting equation. About 99% correlation was achieved for the plot of β0 against the band gap. This linear trend is shown in Fig. 5. An earlier study66 indicates that β0 is inversely related to transition energy (ΔE). Accordingly, VCTO4b, with a minimum transition energy of 2.28 eV, have the highest β0 value of 3278.83 × 10−30 esu. A higher magnitude of β0 and dipole moment is essential for pronounced NLO activity, and the present study clearly illustrates that the D–π–A VCTOs are the best choice of material for NLO applications compared to the reference molecules.
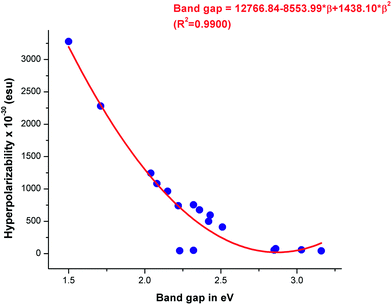 |
| | Fig. 5 Relationship between band gap and hyperpolarizability (β0) from correlation coefficient analysis. | |
3.5 Conducting properties
The ability to transport and inject charge, and the balance of these abilities, is strongly associated with the performance of optoelectronic compounds. Therefore, it is essential to investigate global reactivity descriptors such as ionization potential (IP), electron affinity (EA) and exciton binding energy (Eb) to evaluate the transport ability of these VCTOs (Table 3). The IPs and EAs can be expressed through the HOMO and LUMO orbital energies according to Koopman's theorem.58 It is well known that in OLEDs, a lower IP leads to the easy injection of holes from the hole transport layer (HTL), while a higher EA paves the way for the easy injection of electrons from the electron transport layer (ETL). From Table 3, it is clear that the reference VCTOs (VCTO1, 2 & 3) show higher IPs than the designed VCTOs. The increasing order of IPs in the reference VCTOs is as follows: VCTO1 < VCTO2 < VCTO3. This shows that these VCTOs have poor hole transport character. However, all the designed VCTOs were found to show lower IPs than the reference molecules. Among the D–π–A molecules, all the carboxylic acid derivatives were found to have lower IPs than their corresponding cyano and nitro derivatives. The calculated EA values suggest that carboxylic acid derivatives have lower EAs than the designed VCTOs. This shows that these carboxylic acid derivatives are good hole transport materials. It is important to note that VCTO4ew has a higher EA value than that of VCTO4er. This shows that the EW group improves the electron transporting character; meanwhile, VCTOer has the lowest IP value among all the VCTOs, suggesting that the ER group increases the hole transport properties. Thus, the donor and acceptor moieties alter the IPs and EAs significantly.
Table 3 Calculated ionization potential (IP), electron affinity (EA) and exciton binding energy (Eb) at the B3LYP/6-31g(d) level for VCTOs in eV
| Molecules |
IP |
EA |
Eb |
| VCTO 1 |
5.51 |
3.19 |
−0.10 |
| VCTO1a |
4.67 |
2.31 |
−0.36 |
| VCTO1b |
4.72 |
2.64 |
−0.50 |
| VCTO1c |
4.63 |
2.20 |
−0.29 |
| VCTO2 |
5.64 |
3.40 |
−0.10 |
| VCTO2a |
4.90 |
2.58 |
−0.25 |
| VCTO2b |
4.98 |
2.83 |
−0.35 |
| VCTO2c |
4.86 |
2.43 |
−0.22 |
| VCTO3 |
7.11 |
4.08 |
−0.11 |
| VCTO3a |
5.31 |
2.45 |
0.13 |
| VCTO3b |
5.14 |
2.27 |
0.10 |
| VCTO3c |
4.73 |
1.5 |
0.08 |
| VCTO4ew |
5.13 |
2.91 |
−0.45 |
| VCTO4a |
4.49 |
2.78 |
−0.67 |
| VCTO4b |
4.53 |
3.02 |
−0.78 |
| VCTO4c |
4.41 |
2.36 |
−0.55 |
| VCTO4er |
4.31 |
1.80 |
−0.29 |
Exciton binding energy (Eb) is used to estimate the energy conversion efficiency of a molecule. Hence, the exciton binding energy has been calculated as reported earlier.67 The Eb values reported in Table 3 suggest that the reference VCTOs have nearly the same values, −0.10, −0.10, and −0.11 eV higher than those of the designed VCTOs, respectively. This indicates that D–π–A type molecules are found to have better energy conversion efficiencies than A–π–A type molecules. The energy conversion efficiencies are in good agreement with the calculated FMO energies. For instance, the narrow band gap VCTO4b has the lowest Eb of −0.78 eV, and the wide band gap VCTO3s have higher Eb values. This shows that the lower the HOMO–LUMO gap, the lower the Eb and the higher the energy conversion efficiency.
3.6 Charge analysis
NBO charges are useful to identify the distribution of charge in a molecule.61 Moreover, the charge transport ability of a molecule depends on the magnitude of the charges on the different segments of a molecule. Therefore, NBO charges have been calculated on the donor and acceptor moieties of the VCTOs (SIF6) and the results are shown in Fig. 6 and SIT3. Before discussing the results, it is important to recall that a good charge transport molecule should have more positive charge on the donor moiety which can be readily available for donation. Similarly, the acceptor moiety should accept the donated charges and become more negative. This facilitates the transportation of charge throughout the molecule. From Fig. 6, it is clear that all the donors have more positive charges. Especially, VCTO3a has the most positive charge (+0.336) among all the VCTOs. As expected, the acceptors have more negative charges (from −0.151 to −0.692). D–π–A molecules have especially high positive and negative charges on either end. This suggests that the charge is readily moved from donor to acceptor, thereby causing these molecules to have high charge transport character. At the same time, VCTO4er and VCTO4ew have positive and negative charges on either end, respectively. This shows that these two molecules have relatively less charge transport character than the other VCTO4s.
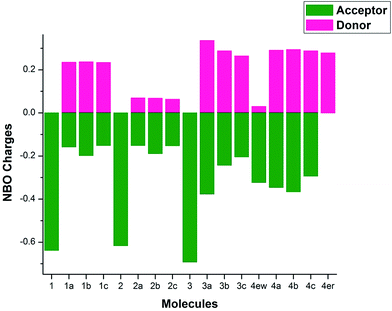 |
| | Fig. 6 NBO charges of VCTOs calculated on donor and acceptor moieties. | |
3.7 Electronic absorption spectra
For a molecule to display good NLO properties, it has to show good absorption and emission properties.54 TDDFT calculations have been performed to compute the excited state properties68–71 and to obtain the electronic vertical singlet excitation energies of the VCTOs in the gas phase. In the literature, different functionals have been reported for the calculation of the absorption spectra of organic molecules.72–74 Therefore, it is essential to use an appropriate functional that gives accurate predictions of the absorption spectra. In order to identify the most reliable method to predict the absorption spectra of these VCTO derivatives, different functionals (B3LYP, B1LYP, CAM-B3LYP, PBE and MO62X) have been tested by comparing the predicted spectra with the available experimental results (SIT4). From the computed results, it is clear that the λmax estimated at the MO62X/6-31g(d) level agrees well with the experimentally observed absorption maxima (see Table SIT4†). Therefore, the M062X functional was used for TDDFT calculations throughout the study.
The calculated vertical transition energies (ΔE), oscillator strengths (f), and transition assignments and their percentages of various configurations to the excitations are summarized in Table 4. The λmax value displayed in Table 4 is the most intense band that can be assigned to the S0 → S1 transition. The computed absorption maxima for all these VCTOs lie in the range of 360–545 nm, which implies that their emissions will be at still longer wavelengths, making them suitable for optoelectronic applications. From the table it is clear that the absorption of these systems can be shifted to longer or shorter wavelengths by substituting ER and EW groups in the skeleton. As expected, the VCTO with the lowest band gap, VCTO4b, has the longest absorption wavelength (545 nm). This arises due to the contributions of 64% & 19% from the HOMO → LUMO and HOMO − 1 → LUMO transitions, respectively. It is evident from the data presented in Table 4 that the VCTO3s have predominant HOMO → LUMO contributions (>97%) for their intense band, which is due to π–π* transitions. However, other VCTOs (the VCTO1, 2 & 4 groups of molecules) were found to show both π–π* transitions and intramolecular charge transfer character. In the VCTO2s, all the molecules were found to have absorption wavelengths of more than 400 nm, with VCTO2b and VCTO2 having the highest and lowest λmax respectively. The substituent EW groups had a larger influence on the absorption spectra than the ER groups. For instance, VCTO4ew has a λmax of 476 nm, while VCTO4er has a λmax of 444 nm.
Table 4 Absorption maxima (λmax), electronic transition energies (ΔE), oscillator strengths (f), light harvesting efficiencies (LHE) and transition assignments of VCTOs in the gas phase from TDDFT calculations at the MO62X/6-31g(d) level
| Molecules |
λmax (gas phase) |
f |
Transition assignments |
| nm |
eV |
| VCTO1 |
404 |
3.07 |
2.74 |
H → L (85%) |
| VCTO1a |
361 |
3.44 |
2.39 |
H → L (62%) |
| VCTO1b |
462 |
2.69 |
2.04 |
H → L (55%) |
| H − 1 → L (20%) |
| H → L + 1 (18%) |
| VCTO1c |
442 |
2.81 |
2.14 |
H → L (66%) |
| VCTO2 |
415 |
2.99 |
2.96 |
H → L (87%) |
| VCTO2a |
468 |
2.65 |
2.29 |
H → L (76%) |
| VCTO2b |
475 |
2.61 |
2.20 |
H → L (68%) |
| VCTO2c |
455 |
2.73 |
2.30 |
H → L (77%) |
| VCTO3 |
371 |
3.35 |
1.13 |
H → L (99%) |
| VCTO3a |
412 |
3.01 |
0.89 |
H → L (99%) |
| VCTO3b |
399 |
3.11 |
0.94 |
H → L (97%) |
| VCTO3c |
373 |
3.32 |
0.72 |
H → L (98%) |
| VCTO4ew |
476 |
2.60 |
1.98 |
H → L (74) |
| VCTO4a |
522 |
2.38 |
1.89 |
H → L (60%) |
| H − 1 → L (21%) |
| VCTO4b |
545 |
2.28 |
1.68 |
H → L (64%) |
| H − 1 → L (19%) |
| VCTO4c |
476 |
2.6 |
2.14 |
H → L (53%) |
| H − 1 → L (21%) |
| H → L + 1 (20%) |
| VCTO4er |
444 |
2.80 |
2.00 |
H → L (74%) |
3.8 Effect of solvent on UV absorption spectra
The solvent effects on the absorption spectra of the VCTOs was investigated by performing PCM calculations with dichloromethane as the solvent. The PCM calculations have been identified as the most successful model for describing solvent effects in DFT and TDDFT calculations.75,76 The ten lowest lying excited states were calculated for the VCTOs, and their important excitation wavelengths, along with their oscillator strengths, are shown in Table 5. It is interesting to note that the calculated absorption maxima of the experimentally reported VCTOs are in good agreement with the experimental report.23 The measured results show that the molecules show a shift in their absorption spectra upon solvation (15–117 nm). Especially, VCTO2 was found to show a shift from 415 nm to 532 nm. However, solvation has little effect on VCTO2a and 2c, which have shifts of only 15 nm upon solvation. The HOMO → LUMO configurations of VCTO1 contribute 83% to the absorption at 513 nm, which is 109 nm higher than that in the gas phase. The transition arising from S0 → S1 is the most intense band in all the VCTOs. All the VCTOs were found to show positive solvatochromism; however, the overall trend in the absorption remains the same in both the gas and solvent phases. Light Harvesting Efficiency (LHE), another interesting property, was calculated and the results are given in Table 5. It is important to note that the LHE varies from 0.85 to 0.99, which is closer to 1. This shows that all the VCTOs are found to have good light harvesting character.
Table 5 Absorption maxima (λmax), electronic transition energies (ΔE), oscillator strength (f) and transition assignments of VCTOs in DCM medium from TDDFT calculations at the MO62X/6-31g(d) level
| Molecules |
λmax (DCM) |
f |
LHE |
Transition assignments |
| nm |
eV |
| VCTO1 |
513 (527) |
2.42 |
2.64 |
0.997 |
H → L (83%) |
| H − 1 → L + 1 (11%) |
| VCTO1a |
455 |
2.72 |
2.31 |
0.995 |
H → L (58%) |
| H − 1 → L (20%) |
| H → L + 1 (15%) |
| VCTO1b |
481 |
2.58 |
2.22 |
0.994 |
H → L (48%) |
| H − 1 → L (24%) |
| H → L + 1 (19%) |
| VCTO1c |
456 |
2.72 |
2.30 |
0.995 |
H → L (61%) |
| H − 1 → L (19%) |
| H → L + 1 (14%) |
| VCTO2 |
532 (530) |
2.33 |
2.81 |
0.998 |
H → L (85%) |
| H − 1 → L + 1 (10%) |
| VCTO2a |
483 |
2.57 |
2.48 |
0.996 |
H → L (76%) |
| H → L + 1 (9%) |
| H − 1 → L (8%) |
| VCTO2b |
495 |
2.5 |
2.40 |
0.996 |
H → L (65%) |
| H → L + 1 (15%) |
| H − 1 → L (15%) |
| VCTO2c |
470 |
2.64 |
2.47 |
0.996 |
H → L (76%) |
| H → L + 1 (9%) |
| H − 1 → L (8%) |
| VCTO3 |
394 (406) |
3.14 |
0.91 |
0.878 |
H → L (99%) |
| VCTO3a |
456 |
2.72 |
1.03 |
0.907 |
H → L (98%) |
| VCTO3b |
449 |
2.76 |
1.07 |
0.916 |
H → L (96%) |
| VCTO3c |
402 |
3.08 |
0.83 |
0.852 |
H → L (97%) |
| VCTO4ew |
499 |
2.49 |
2.11 |
0.992 |
H → L (73%) |
| H − 1 → L (15%) |
| H → L + 1 (8%) |
| VCTO4a |
544 |
2.28 |
2.10 |
0.992 |
H → L (55%) |
| H − 1 → L (25%) |
| H → L + 1 (14%) |
| VCTO4b |
576 |
2.15 |
1.84 |
0.985 |
H → L (59%) |
| H − 1 → L (24%) |
| H → L + 1 (8%) |
| VCTO4c |
496 |
2.50 |
2.29 |
0.994 |
H → L (48%) |
| H − 1 → L (23%) |
| H → L + 1 (21%) |
| VCTO4er |
461 |
2.69 |
2.09 |
0.991 |
H → L (72%) |
| H − 1 → L (11%) |
| H → L + 1 (9%) |
3.9 NBO analysis
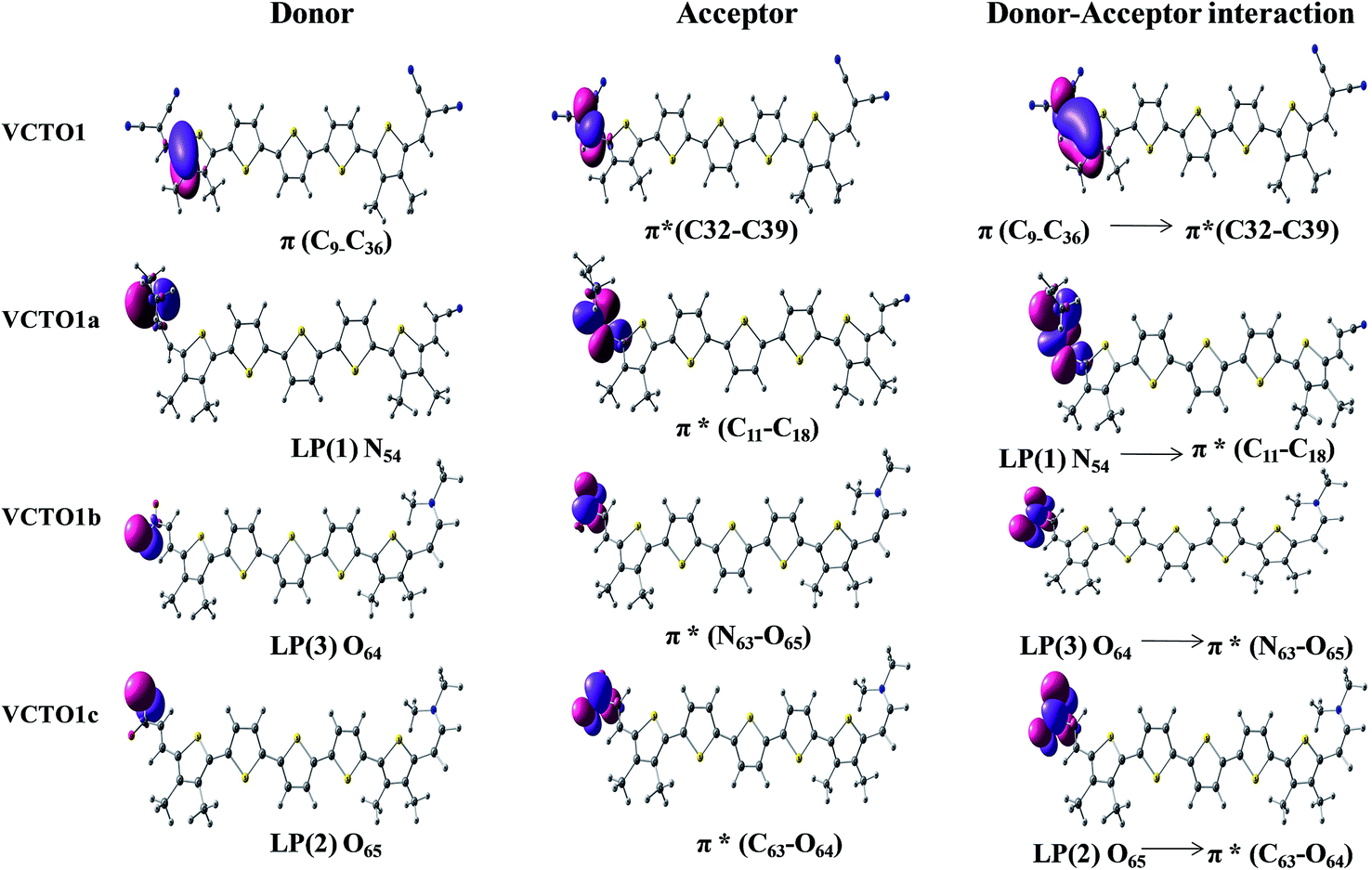
NBO analysis is a powerful tool that reveals the nature of the electronic interactions and electronic structure properties, which are solely responsible for deciding the chemical characteristics of molecules.27,54,61 These properties are orbital energy, % electron density (%ED), stabilization energy (E2) and the energy splitting (Ej − Ei) between donor and acceptor orbitals. The energy gap between the bonding orbital and the antibonding orbital decides the feasibility of interaction between filled donor and acceptor moieties. The %ED of the donors and acceptors involved in hyperconjugative interaction, the orbital energies, and the stabilization energies have been computed using a second order perturbation approach61 at the B3LYP/6-31g (d) level, and the results are displayed in Table 6. The interactions between the occupied and unoccupied levels of the VCTO1s are depicted in Fig. 7, and the same interactions for the other systems are collected in SIF7–SIF10.
Table 6 Stable NBO donor–acceptor interactions of VCTOs with its occupancy, %ED, and orbital energy obtained from B3LYP/6-31g(d) calculations. E(2) is the energy of hyperconjugative interaction; ED is the electron density; Ej − Ei is the energy difference between the donor (i) and acceptor (j) NBO orbitals
| Molecules |
Donor (i) |
ED (i) (e) |
%ED polarization |
Orbital energy (au) |
Acceptor (j) |
ED (j) (e) |
%ED polarization |
Orbital energy (au) |
E(2) kcal mol−1 |
Ej − Ei (au) |
| VCTO1 |
π(C9–C36) |
1.7076 |
57.10C9–42.90C36 |
−0.2516 |
π*(C32–C39) |
0.2833 |
59.07C32–40.93C39 |
0.0462 |
26.50 |
0.28 |
| VCTO1a |
LP(1) N54 |
1.7016 |
— |
−0.2408 |
π*(C11–C18) |
0.2437 |
44C11–56C18 |
0.0544 |
39.86 |
0.30 |
| VCTO1b |
LP(3) O64 |
1.4822 |
— |
−0.2549 |
π*(N63–O65) |
0.6520 |
59.96N63–40.04O65 |
−0.1153 |
154.88 |
0.14 |
| VCTO1c |
LP(2) O65 |
1.8240 |
— |
−0.3181 |
π*(C63–O64) |
0.2910 |
70.35C63–29.65O64 |
0.0188 |
44.79 |
0.34 |
| VCTO2 |
π(C31C39) |
1.7173 |
56.38C31–43.62C39 |
−0.2971 |
π*(C34–O13) |
0.2787 |
58.85C34–41.15O13 |
−0.0079 |
26.10 |
0.29 |
| VCTO2a |
LP(2) S2 |
1.6710 |
— |
−0.2607 |
π*(C13–C14) |
0.3685 |
48.44C13–51.56C14 |
0.0049 |
21.31 |
0.27 |
| VCTO2b |
LP(3) O53 |
1.4781 |
— |
−0.2578 |
π*(N51–O52) |
0.6508 |
59.88N51–40.12O52 |
−0.1185 |
156.27 |
0.14 |
| VCTO2c |
LP (2) O52 |
1.8232 |
— |
−0.3210 |
π*(C51–O53) |
0.2880 |
70.21C51–29.79O53 |
0.0161 |
44.92 |
0.34 |
| VCTO3 |
LP(2) S1 |
1.6041 |
— |
−0.2911 |
π*(C6–C7) |
0.3856 |
45.65C6–54.35C7 |
−0.0385 |
23.07 |
0.25 |
| VCTO3a |
LP(1) O24 |
1.6634 |
— |
−0.2718 |
π*(C2–C11) |
0.2844 |
42.66C2–57.34C11 |
0.0151 |
49.71 |
0.29 |
| VCTO3b |
LP(3) O24 |
1.4942 |
— |
−0.2491 |
π*(N23–O25) |
0.6712 |
60.42N23–39.58O25 |
−0.1076 |
146.51 |
0.14 |
| VCTO3c |
LP(2) O25 |
1.8289 |
— |
−0.3103 |
π*(C23–O24) |
0.3090 |
70.96C23–29.04O24 |
0.0237 |
43.86 |
0.33 |
| VCTO4ew |
π(C8–C15) |
1.7050 |
57.23C8−42.77C15 |
−0.2819 |
π*(C11–C18) |
0.2914 |
59.26C11–40.74C18 |
0.0014 |
27.02 |
0.28 |
| VCTO4a |
LP(1) N59 |
1.7564 |
— |
−0.2575 |
π*(C32–C39) |
0.3297 |
43.13C32–56.87C39 |
0.0387 |
27.94 |
0.30 |
| VCTO4b |
LP(3) O77 |
1.4807 |
— |
−0.2701 |
π*(N75–O76) |
0.6335 |
59.01N75–40.99O76 |
−0.1232 |
148.24 |
0.15 |
| VCTO4c |
LP(2) O74 |
1.8092 |
— |
−0.317 |
π*(C72–O73) |
0.2862 |
70.85C72–29.15O73 |
0.0133 |
48.59 |
0.33 |
| VCTO4er |
LP(1) N55 |
1.7606 |
— |
−0.2516 |
π*(C28–C35) |
0.3255 |
43.23C28–56.77C35 |
0.0462 |
27.62 |
0.30 |
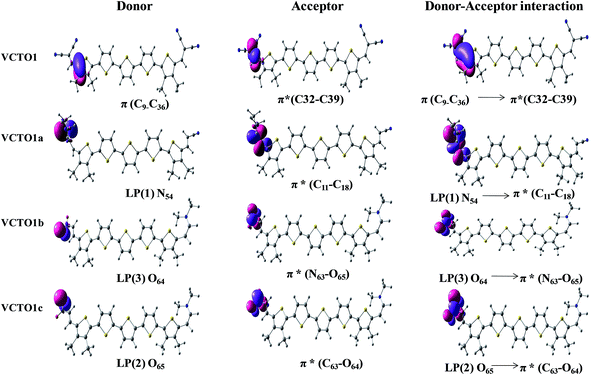 |
| | Fig. 7 Illustration of second order perturbation interactions in VCTO1s from NBO analysis. | |
As seen from the table, the stabilization energies are high for VCTOs1b, 2b, 3b and 4c and increase in the following order: VCTO3b > VCTO4b > VCTO1b > VCTO2b. The calculated % of electron density distribution (ED) on the non-Lewis moieties in VCTO2a is 48.44 (C13)–51.56 (C14). Though several types of electronic interactions are present between the occupied and unoccupied levels, the most dominant stabilization comes from the n → π* and π → π* transitions in the VCTOs. The lone pairs (LP) of N, S and O donate electrons to the antibonding π*C–C, π*C–O, and π*N–O orbitals. The second order stabilization (39.86 kJ mol−1) in VCTO1a is due to the lone pair localized on N54 and π*C11–C18. The computed second order perturbation analysis conducted on the VCTOs clearly conveys the presence of the most stabilizing orbital interaction between the donor and acceptor fragments. Especially, all the nitro derivative VCTOs were found to show larger second order perturbation interactions than the other VCTOs (Table 6).
The present studies show that 9 out of 17 of the designed VCTOs have promising optoelectronic properties compared to their corresponding reference VCTOs. Among the designed VCTOs, VCTO4b was found to show excellent optoelectronic properties, with a low band gap (1.50 eV), high wavelength of absorption (576 nm), and remarkable NLO properties (β0 = 3278.83 esu); thus, this VCTO is the most promising for future optoelectronic applications.
4. Conclusions
In total, 17 VCTOs, 3 of them reported by Peter Baurele et al. and 14 newly designed here, have been studied using DFT/TDDFT methods in order to identify the candidates with the best optoelectronic properties. The computed results show that experimentally synthesized, acceptor–acceptor type VCTOs have low band gaps, and the designed candidates have high wavelengths of absorption. VCTO4b was found to be the best candidate, with a low band gap, whereas VCTO3c was found to be the poorest candidate. Furthermore, VCTO4b possesses a high wavelength of absorption, high β0 value and high dipole moment. HOMO–LUMO analysis reveals that the –NO2 group plays a major role in stabilizing the LUMO compared to other electron acceptor moieties (–CN & –COOH). The reference VCTOs showed π → π* transitions, while the designed VCTOs displayed n → π* transitions with IMCT character. The exciton binding energy was lower for the designed VCTOs, which exhibit much greater light harvesting efficiencies than the reference molecules. The electronic stabilization of the molecules was studied by NBO analysis. Furthermore, this study reveals that the reference VCTOs can be better electron transport materials, while the designed VCTOs are better hole transport materials, which indicates that donor–acceptor substitution in the skeleton can convert an electron transport material into a hole transport material. Overall, the present study rationalizes that the optical properties of VCTOs can be tuned by suitable substitution to achieve remarkable optoelectronic properties.
Acknowledgements
RVS thanks University Grants Commission (UGC), India, for financial support in the form of Junior and Senior Research Fellowships in Maulana Azad National Fellowship (Ref. no. F.40-17(C/M)/2009(SA-III/MANF). PV thanks UGC for Emeritus Fellowship (F.6-6/2012-2013/EMERITUS-2012-2013-GEN-1171/(SA-II). We thank the reviewers for their helpful suggestions to improve the clarity of this work.
References
- C.-J. Huang, J.-C. Ke, W.-R. Chen, T.-H. Meen and C.-F. Yang, Sol. Energy Mater. Sol. Cells, 2011, 95, 3460–3464 CrossRef CAS PubMed.
- Y. Li, T. Pullerits, M. Zhao and M. Sun, J. Phys. Chem. C, 2011, 115, 21865–21873 CAS.
- G. Paramaguru, R. V. Solomon, S. Jagadeeswari, P. Venuvanalingam and R. Renganathan, Eur. J. Org. Chem., 2014, 2014, 753–766 CrossRef CAS PubMed.
- E. Sengupta, A. L. Domanski, S. A. Weber, M. B. Untch, H.-J. R. Butt, T. Sauermann, H. J. Egelhaaf and R. D. Berger, J. Phys. Chem. C, 2011, 115, 19994–20001 CAS.
- H. Tachikawa and H. Kawabata, J. Organomet. Chem., 2012, 720, 60–65 CrossRef CAS PubMed.
- M. R. Raj, S. Anandan, R. V. Solomon, P. Venuvanalingam, S. S. K. Iyer and M. Ashokkumar, J. Photochem. Photobiol., A, 2012, 247, 52–62 CrossRef CAS PubMed.
- Y. Zhou, J. W. Shim, C. Fuentes-Hernandez, T. M. Khan and B. Kippelen, Thin Solid Films, 2014, 554, 54–57 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe and J. Kim, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- Z. Hu, C.-A. Tao, F. Wang, X. Zou and J. Wang, J. Mater. Chem. C, 2015, 3, 211–216 RSC.
- C. H. Hendon, D. Tiana, M. Fontecave, C. M. Sanchez, L. D'arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed.
- Y. Zhou, Z. Wang, P. Yang, X. Zu and F. Gao, J. Mater. Chem., 2012, 22, 16964–16970 RSC.
- L.-M. Yang and R. Pushpa, J. Mater. Chem. C, 2014, 2, 2404–2416 RSC.
- E. Klontzas, E. Tylianakis and G. E. Froudakis, Nano Lett., 2010, 10, 452–454 CrossRef CAS PubMed.
- M. D. Allendorf, A. Schwartzberg, V. Stavila and A. Alec Talin, Chem.–Eur. J., 2011, 17, 11372–11388 CrossRef CAS PubMed.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- M. Klimakow, P. Klobes, K. Rademann and F. Emmerling, Microporous Mesoporous Mater., 2012, 154, 113–118 CrossRef CAS PubMed.
- C. K. Brozek and M. Dinca, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed.
- N. Nijem, H. Wu, P. Canepa, A. Marti, K. J. Balkus Jr, T. Thonhauser, J. Li and Y. J. Chabal, J. Am. Chem. Soc., 2012, 134, 15201–15204 CrossRef CAS PubMed.
- R. Fitzner, E. Mena-Osteritz, A. Mishra, G. Schulz, E. Reinold, M. Weil, C. Körner, H. Ziehlke, C. Elschner, K. Leo, M. Riede, M. Pfeiffer, C. Uhrich and P. Bäuerle, J. Am. Chem. Soc., 2012, 134, 11064–11067 CrossRef CAS PubMed.
- L. Dou, J. You, J. Yang, C.-C. Chen, Y. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Nat. Photonics, 2012, 6, 180–185 CrossRef CAS PubMed.
- A. Diaz, K. K. Kanazawa and G. P. Gardini, J. Chem. Soc., Chem. Commun., 1979, 635–636 RSC.
- J. Roncali, Chem. Rev., 1992, 92, 711–738 CrossRef CAS.
- R. V. Solomon, R. Jagadeesan, S. A. Vedha and P. Venuvanalingam, Dyes Pigm., 2014, 100, 261–268 CrossRef CAS PubMed.
- G. Tourillon and F. Garnier, J. Electrochem. Soc., 1983, 130, 2042–2044 CrossRef CAS PubMed.
- R. J. Waltman, J. Bargon and A. Diaz, J. Phys. Chem., 1983, 87, 1459–1463 CrossRef CAS.
- S. Glenis, M. Benz, E. LeGoff, J. Schindler, C. Kannewurf and M. Kanatzidis, J. Am. Chem. Soc., 1993, 115, 12519–12525 CrossRef CAS.
- E. Maarouf, D. Billaud and E. Hannecart, Mater. Res. Bull., 1994, 29, 637–643 CrossRef CAS.
- T.-Y. Chu, S. Alem, P. G. Verly, S. Wakim, J. Lu, Y. Tao, S. Beaupré, M. Leclerc, F. Bélanger and D. Désilets, Appl. Phys. Lett., 2009, 95, 063304 CrossRef PubMed.
- M. Delamar, P.-C. Lacaze, J.-Y. Dumousseau and J.-E. Dubois, Electrochim. Acta, 1982, 27, 61–65 CrossRef CAS.
- J. Rault-Berthelot, L. Angely, J. Delaunay and J. Simonet, New J. Chem., 1987, 11, 487–494 CAS.
- A. Bongini, G. Barbarella, L. Favaretto, G. Sotgiu, M. Zambianchi and D. Casarini, Tetrahedron, 2002, 58, 10151–10158 CrossRef CAS.
- J. Brédas, J. Cornil, F. Meyers, D. Beljonne, T. Skotheim, R. Elsenbaumer and J. Reynolds, Handb. Conduct. Polym., 1998, 1–26 Search PubMed.
- D. Fichou, Handbook of oligo-and polythiophenes, John Wiley & Sons, 2008 Search PubMed.
- F. Garnier, Acc. Chem. Res., 1999, 32, 209–215 CrossRef CAS.
- K. Mullen and G. Wegner, Electronic materials, Wiley, 2008 Search PubMed.
- J. Rogers and H. Katz, J. Mater. Chem., 1999, 9, 1895–1904 RSC.
- A. Alberti, L. Favaretto, G. Seconi and G. F. Pedulli, J. Chem. Soc., Perkin Trans. 2, 1990, 931–935 RSC.
- Y. Cao, D. Guo, M. Pang and R. Qian, Synth. Met., 1987, 18, 189–194 CrossRef CAS.
- J. V. Caspar, V. Ramamurthy and D. R. Corbin, J. Am. Chem. Soc., 1991, 113, 600–610 CrossRef CAS.
- D. Fichou, G. Horowitz and F. Garnier, Synth. Met., 1990, 39, 125–131 CrossRef CAS.
- H. Fujimoto, U. Nagashima, H. Inokuchi, K. Seki, Y. Cao, H. Nakahara, J. Nakayama, M. Hoshino and K. Fukuda, J. Chem. Phys., 1990, 92, 4077–4092 CrossRef CAS PubMed.
- D. Jones, M. Guerra, L. Favaretto, A. Modelli, M. Fabrizio and G. Distefano, J. Phys. Chem., 1990, 94, 5761–5766 CrossRef CAS.
- G. Zerbi, B. Chierichetti and O. Ingänas, J. Chem. Phys., 1991, 94, 4637–4645 CrossRef CAS PubMed.
- U. Schoeler, K. Tews and H. Kuhn, J. Chem. Phys., 1974, 61, 5009–5016 CrossRef CAS PubMed.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern and J. A. Pople, Chem. Phys. Lett., 1997, 270, 419–426 CrossRef CAS.
- E. Kleinpeter and B. A. Stamboliyska, J. Org. Chem., 2008, 73, 8250–8255 CrossRef CAS PubMed.
- R. V. Solomon, P. Veerapandian, S. A. Vedha and P. Venuvanalingam, J. Phys. Chem. A, 2012, 116, 4667–4677 CrossRef CAS PubMed.
- K. S. Thanthiriwatte and K. Nalin de Silva, J. Mol. Struct.: THEOCHEM, 2002, 617, 169–175 CrossRef CAS.
- R. Improta, V. Barone, G. Scalmani and M. J. Frisch, J. Chem. Phys., 2006, 125, 054103 CrossRef PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- T. Koopmans, Physica, 1934, 1, 104–113 CrossRef.
- E. Glendening, A. Reed, J. Carpenter and F. Weinhold, NBO version, 1998, vol. 3 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. Weinhold, J. Comput. Chem., 2012, 33, 2363–2379 CrossRef CAS PubMed.
- M. J. T. Frisch, G. W. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. G. Pearson, in Chemical hardness, Springer, 1993, pp. 1–10 Search PubMed.
- R. V. Solomon, A. P. Bella, S. A. Vedha and P. Venuvanalingam, Phys. Chem. Chem. Phys., 2012, 14, 14229–14237 RSC.
- D. Kleinman, Phys. Rev., 1962, 126, 1977 CrossRef CAS.
- M. R. S. A. Janjua, M. U. Khan, B. Bashir, M. A. Iqbal, Y. Song, S. A. R. Naqvi and Z. A. Khan, Comput. Theor. Chem., 2012, 994, 34–40 CrossRef CAS PubMed.
- R. Nithya and K. Senthilkumar, Phys. Chem. Chem. Phys., 2014, 16, 21496–21505 RSC.
- C. M. Aikens, S. Li and G. C. Schatz, J. Phys. Chem. C, 2008, 112, 11272–11279 CAS.
- E. Gross and W. Kohn, Adv. Quantum Chem., 1990, 21, 255–291 CAS.
- J. Kulhánek, F. Bures, A. Wojciechowski, M. Makowska-Janusik, E. Gondek and I. Kityk, J. Phys. Chem. A, 2010, 114, 9440–9446 CrossRef PubMed.
- M. Makowska-Janusik, I. Kityk, J. Kulhanek and F. Bures, J. Phys. Chem. A, 2011, 115, 12251–12258 CrossRef CAS PubMed.
- P. J. Aittala, O. Cramariuc, T. I. Hukka, M. Vasilescu, R. Bandula and H. Lemmetyinen, J. Phys. Chem. A, 2010, 114, 7094–7101 CrossRef CAS PubMed.
- M. J. G. Peach, C. R. Le Sueur, K. Ruud, M. Guillaume and D. J. Tozer, Phys. Chem. Chem. Phys., 2009, 11, 4465–4470 RSC.
- I. Petsalakis, D. Georgiadou, M. Vasilopoulou, G. Pistolis, D. Dimotikali, P. Argitis and G. Theodorakopoulos, J. Phys. Chem. A, 2010, 114, 5580–5587 CrossRef CAS PubMed.
- A. Pedone, J. Bloino, S. Monti, G. Prampolini and V. Barone, Phys. Chem. Chem. Phys., 2009, 12, 1000–1006 RSC.
- V. Barone, J. Bloino, S. Monti, A. Pedone and G. Prampolini, Phys. Chem. Chem. Phys., 2010, 12, 10550–10561 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: XYZ coordinates, bond parameters, NBO analysis, and HOMO–LUMO energies. See DOI: 10.1039/c5ra01466b |
| ‡ Present address: Department of Chemical Engineering, University of South Carolina, Columbia, USA. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.