DOI:
10.1039/C5RA01450F
(Paper)
RSC Adv., 2015,
5, 27140-27153
Stabilization of η3-indenyl compounds by sterically demanding N,N-chelating ligands in the molybdenum coordination sphere†
Received
24th January 2015
, Accepted 11th March 2015
First published on 16th March 2015
Abstract
A series of η3-indenyl molybdenum compounds [(η3-4,7-Me2C9H5)Mo(CO)2(N,NL)Cl] (N,NL = bpy, phen, pyma), isostructural with well-known η3-allyl compounds, was synthesized from the recently established halide synthon [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2]. The low stability of the hexacoordinated η3-indenyl molybdenum species in solution has been overcome by a modification of the chelating ligand. Hence, the dissociation of the compounds bearing ligands with methyl groups beside nitrogen donor atoms (e.g. 6,6′-Me2-bpy, 2,9-Me2-phen; 2,9-Me2-4,7-Ph2-phen) is strongly disfavored due to the steric requirements of the substituents. The considerable discrimination of the pentacoordinated species enables the use of [(η5-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)][BF4] for the assembly of derivatives bearing other halides and pseudohalides in the coordination sphere of molybdenum. The current study further describes some other new indenyl complexes accessible from [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2]. All structural types presented in this experimental study were supported by X-ray crystallographic data.
Introduction
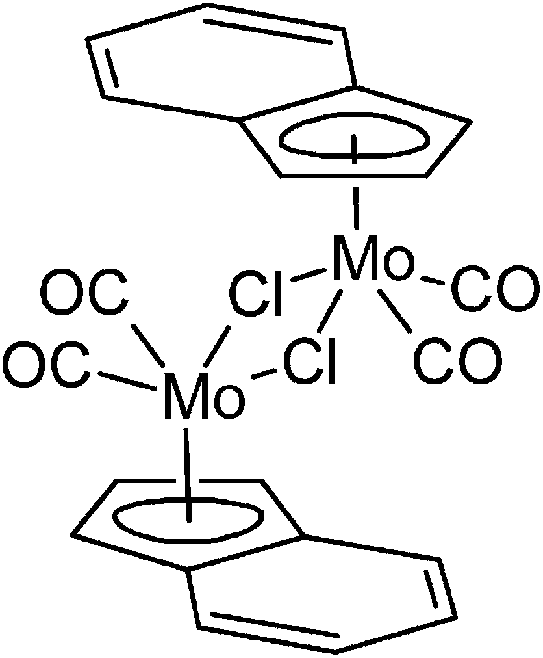
Organometallic molybdenum compounds are currently under comprehensive scrutiny due to their interesting properties. Allyl compounds [(η3-C3H5)Mo(CO)2(N,NL)Cl] (N,NL = N,N-chelating ligand) show a rich coordination chemistry and have found several applications in organic synthesis and catalysis.1–11 Promising cytotoxic properties have been observed for cationic cyclopentadienyl and indenyl compounds [(η5-Cp′)Mo(CO)2(X,XL)][BF4] (Cp′ = Cp = C5H5, Ind = C9H7), where X,XL is N,N-, P,P- or S,S-chelating ligands, and their congeners with substituted π-ligands.12–14 Neutral cyclopentadienyl compounds [(η5-Cp′)Mo(CO)3X] (Cp′ = substituted Cp, X = anionic ligand) are well-established precursors of catalysts for oxidation reactions (e.g. olefin epoxidation or sulfoxidation).15–21 Recently, we have described a synthesis of [{(η5-Ind)Mo(CO)2(μ-Cl)}2] (1; Scheme 1) that was found to be a versatile synthon for various η3-indenyl compounds.22 Furthermore, this compound seems to be a suitable precatalyst for isomerization of α-pinene oxide to campholenic aldehyde23 and a precursor of hydrolytically active species for promoted hydrolysis of phosphoesters.24
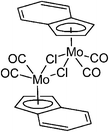 |
| | Scheme 1 Molecular formula of 1. | |
Although the cyclopentadienyl and indenyl molybdenum compounds have often a similar molecular structure, a different reactivity is expected since a replacement of the cyclopentadienyl ligand with the indenyl accelerates reaction rates due to a lower energetic barrier of the haptotropic shift of the π-ligand.25–29 The kinetic “indenyl effect” has been comprehensively scrutinized on various molybdenum compounds by Romão and Calhorda.30–32 For instance, they have recently demonstrated that the indenyl compound [(η5-Ind)Mo(CO)2(κ2-ttcn)][BF4] (ttcn = 1,4,7-trithiacyclononane) undergoes an acid-activated C–S cleavage to give [(η5-Ind)Mo(CO){κ3-(SCH2CH2)2S}][BF4], while the cyclopentadienyl analogue is stable under similar conditions.33 An activation of coordinated ligand was also observed for complexes with η4-bonded spiro[2.4]hepta-4,6-diene. The cyclopentadienyl compound [(η5-Cp){η4-C5H4(CH2)2}Mo(CO)2][BF4] is unusually stable, while the indenyl analogue undergoes a spontaneous and very fast ring-opening to [(η5-Ind)(η5-C5H4CH2-η1-CH2)Mo(CO)][BF4].34,35
The aim of this study is to bring a detail description of the coordination properties of [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2) in attempt to synthesize new η3-indenyl compounds structurally related with the well-known complexes bearing η3-allyl [(η3-C3H5)Mo(CO)2(N,NL)Cl]. It will be shown that a steric congestion of the chelating ligand is critical for the stability of [(η3-4,7-Me2C9H5)Mo(CO)2(N,NL)Cl] and could be used for the control of the indenyl ring slippage. When this work was in progress, the first notes about reactivity of the parent compound 1 with coordinating solvents have appeared in literature23,24 but the detailed description of the coordination behavior is still missing. Our current study deals with species bearing 4,7-dimethyl indenyl ligand mainly due to higher solubility of 2 in organic solvents.22 This substitution pattern usually has only negligible effect on the reactivity due to subtle steric and electronic effects of two methyl groups in the C6-ring.36,37
Results and discussion
Reactions of [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2) with py, bpy, phen and pyma
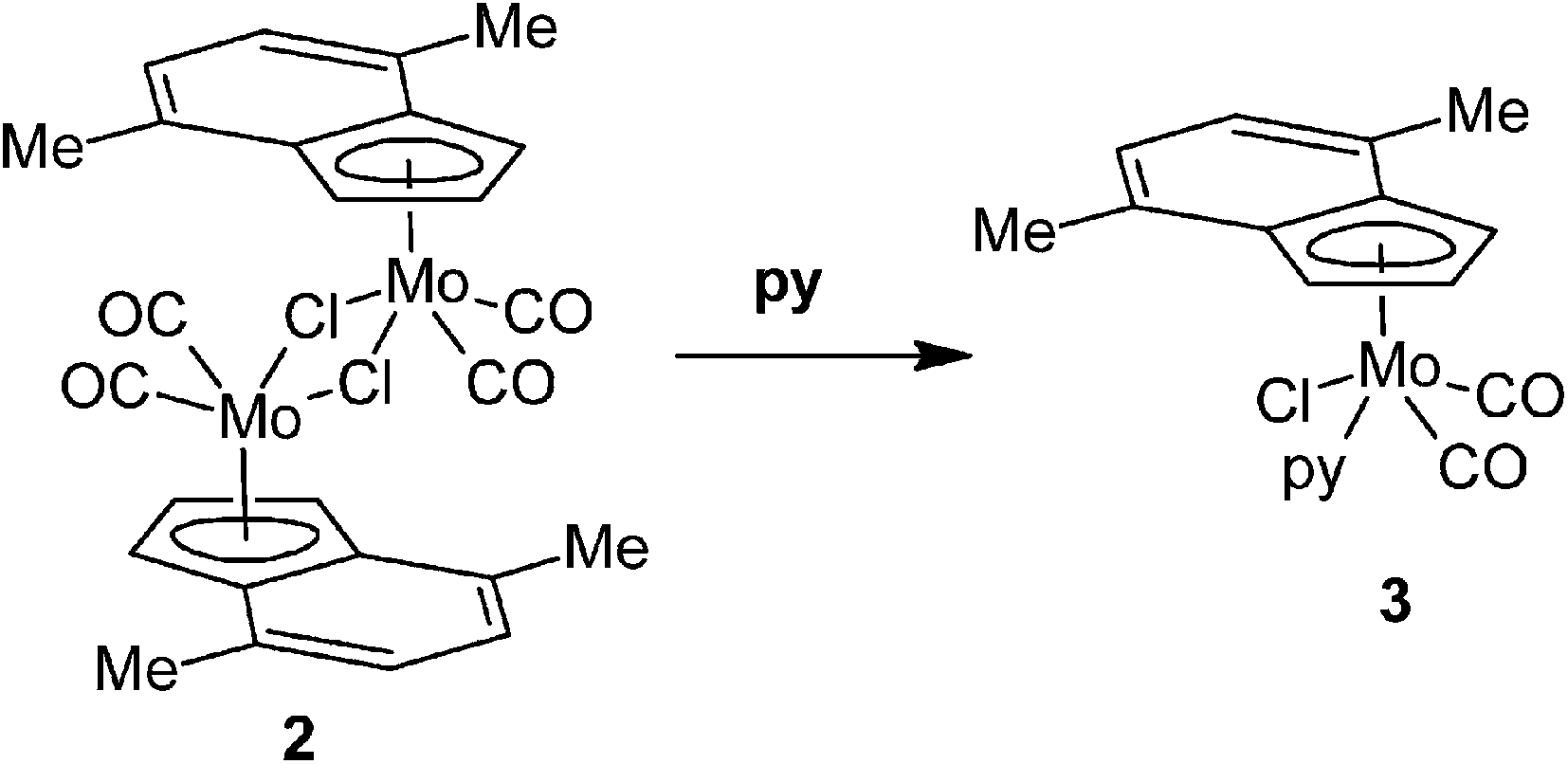
Reaction of [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2) with pyridine (py) gives a monomeric compound with η5-bonded indenyl ligand [(η5-4,7-Me2C9H5)Mo(CO)2(py)Cl] (3), see Scheme 2. The expected η3-indenyl species, which could appear upon coordination of further py molecule to molybdenum, was not detected even when a large excess of the reagent was used. Although the compound 3 is a structural analogue of the previously described cyclopentadienyl compound [(η5-Cp)Mo(CO)2(py)Cl],38 the original route is not suitable for 3 due to a low stability of the indenyl precursor [(η5-Ind)Mo(CO)3Cl] at elevated temperature.39
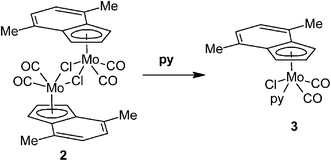 |
| | Scheme 2 Reaction of 2 with pyridine. | |
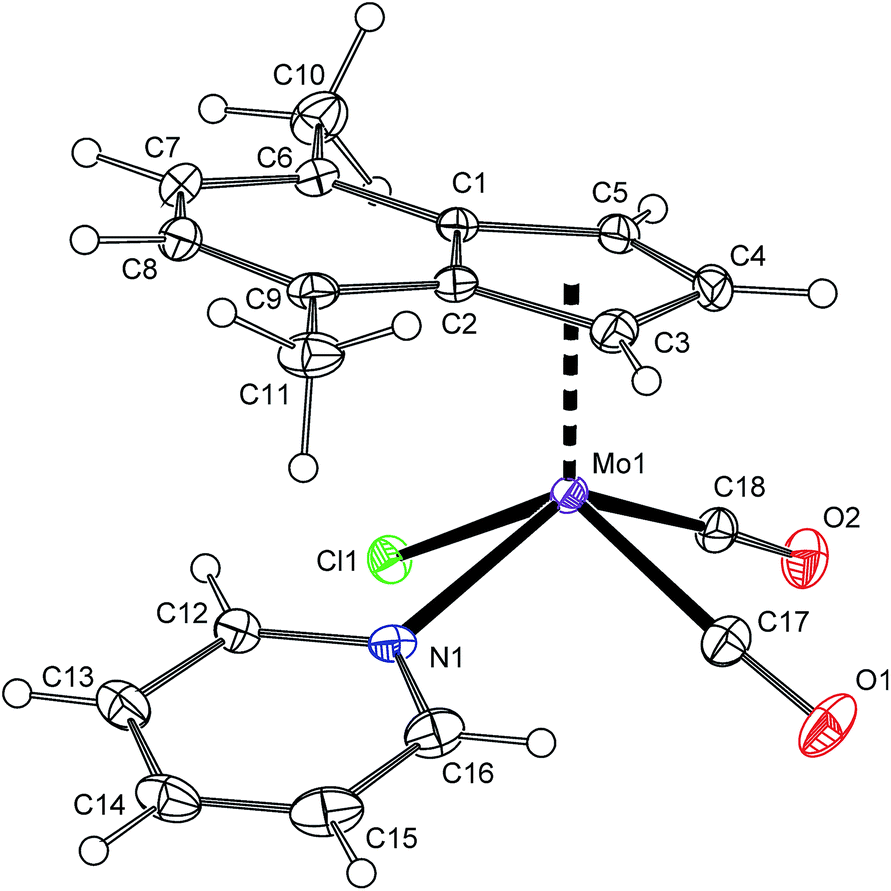
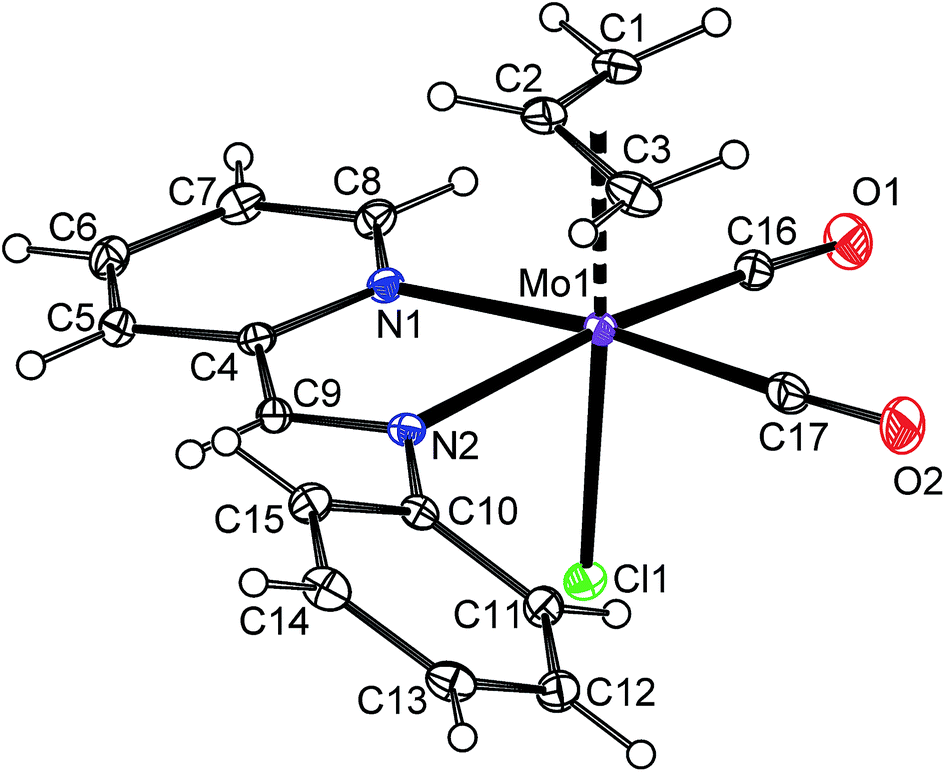
1H NMR spectrum shows the signal of H2 at a high field that is typical for the species with η5-coordinated indenyl ligand.40 The spectrum pattern with magnetically inequivalent protons H1 and H3 imply the C1 point symmetry for the molecule of 3 that is fully in agreement with the solid state structure revealed by single crystal X-ray analysis (Fig. 1 and Table 1). The molecule of 3 has a square pyramidal structure with the η5-bonded indenyl ligand in the apical position. The basal plane is occupied with two cis-coordinated carbonyl ligands, one chloride and the nitrogen donor atom of pyridine. The η5-coordination mode of the indenyl ligand is confirmed by a low value of the envelope fold angle [Ω = 7.4(2)°] and also by Δ(M–C) [0.212(2) Å].
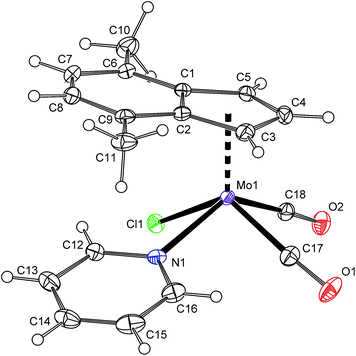 |
| | Fig. 1 ORTEP drawing of [(η5-4,7-Me2C9H5)Mo(CO)2(py)Cl] (3). The labeling for all non-hydrogen atoms is shown. Thermal ellipsoids are drawn at the 30% probability level. | |
Table 1 Geometric parameters of the neutral and cationic pentacoordinated molybdenum speciesa
| |
3
e
|
4b-BF4
|
5b-9·CH2Cl2 |
8-BF4
|
10b-BF4·0.5(Me2-phen·HBF4) |
|
Distances are given in Å; angles and dihedral angles are given in °.
Pl1 is defined by Mo and two nitrogen donor atoms; Pl2 is defined by two nitrogen donor atoms and two adjacent carbon atoms of the chelate ring.
Ω is the envelope fold angle defined for the indenyl ligand as the angle between planes defined by C3, C4 and C5 and that of C1, C2, C3 and C5.41
Δ(M–C) represents the differences in the metal–carbon bonds. It is defined for the indenyl compounds as the difference between the averages of the metal–carbon distances M–C1 and M–C2 and those of M–C3, M–C4, and M–C5.41
Mo–Cl = 2.4998(6) Å; N–Mo–Cl = 82.24(4)°.
|
| Mo–Cg(C5) |
2.0236(9) |
1.9979(12) |
2.004(3) |
1.990(2) |
2.0154(18) |
| Mo–C(CO) |
1.929(2) |
1.960(3) |
1.950(9) |
1.968(5) |
1.945(6) |
| 1.956(2) |
1.970(3) |
1.980(9) |
1.969(4) |
1.957(6) |
| Mo–N |
2.280(2) |
2.190(2) |
2.196(5) |
2.180(4) |
2.244(4) |
| 2.191(2) |
2.208(6) |
2.182(4) |
2.250(4) |
| C(CO)–Mo–C(CO) |
72.71(10) |
74.72(12) |
74.8(4) |
74.3(2) |
69.5(2) |
| N–Mo–N |
— |
72.88(8) |
73.3(2) |
78.93(14) |
74.45(13) |
| Pl1–Pl2b |
— |
7.8(2) |
6.8(4) |
— |
22.4(2) |
|
Ω
c
|
7.4(2) |
5.1(3) |
5.7(8) |
6.7(5) |
6.3(4) |
| Δ(M–C)d |
0.212(2) |
0.149(3) |
0.157(8) |
0.160(5) |
0.178(4) |
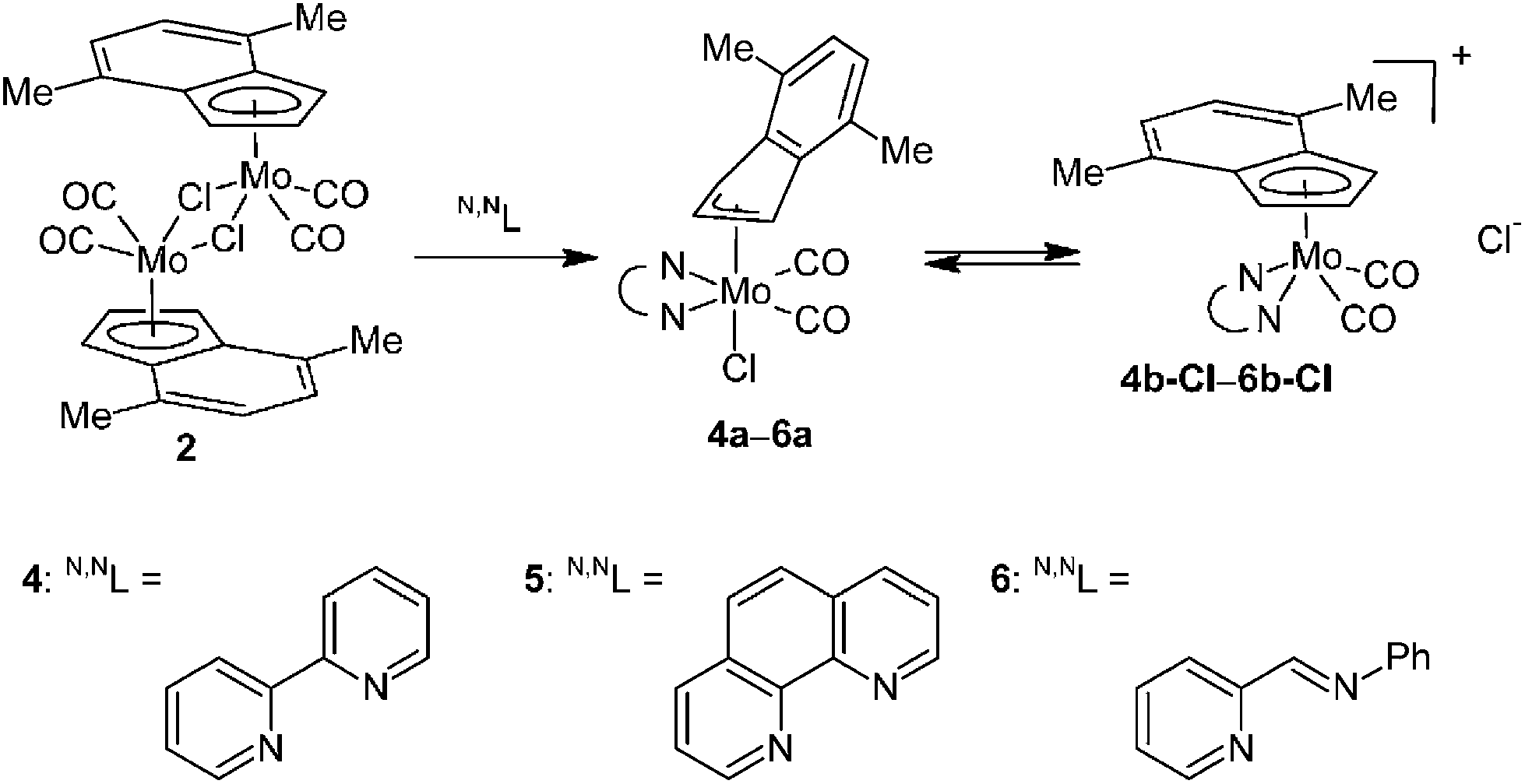
From a mechanistic point of view, the η5–η3 rearrangement of the indenyl ligand is stimulated by a coordination of two 2e donors to molybdenum that could be further enforced by a chelating effect. Nevertheless, this process is further complicated by a coordination or abstraction of chloride as will be documented on the reactions with 2,2′-bipyridine (bpy), 1,10-phenanthroline (phen) and trans-N-(2-pyridylmethylene)aniline (pyma).
Reaction of 2 with two equivalents of bpy, phen and pyma gives, after standard work up, the desired η3-indenyl compounds [(η3-4,7-Me2C9H5)Mo(CO)2(N,NL)Cl] (4a: N,NL = bpy, 5a: N,NL = phen, 6a: N,NL = pyma), see Scheme 3. Infrared spectra of the solid-state samples show two CO stretching bands at low frequencies (Table 2) indicating the proposed neutral structure. Hence, cationic complexes [(η5-Ind)Mo(CO)2L2]+ have much lower electron density on molybdenum atom than neutral compounds with slipped indenyl ring [(η3-Ind)Mo(CO)2L2Cl] as could be documented on the CO stretching band frequencies of the pairs bearing monodentate ligands (cf. data for compounds bearing DMF or MeCN in Table 2).
 |
| | Scheme 3 Reaction of 2 with two equivalents of bpy, phen and pyma. | |
Table 2 Summary of the infrared data for the molybdenum complexesa
| |
ν
a(CO) |
ν
s(CO) |
Ref. |
|
The stretching frequencies are given in cm−1.
|
|
4a
|
1940 |
1862 |
|
|
4b-BF4
|
1970 |
1894 |
|
| [(η5-Ind)Mo(CO)2(bpy)][BF4] |
1974 |
1878 |
13
|
|
5a
|
1919 |
1844 |
|
|
5b-BF4
|
1967 |
1880 |
|
| [(η5-Ind)Mo(CO)2(phen)][BF4] |
1968 |
1874 |
13
|
|
6a
|
1934 |
1855 |
|
| [(η3-Ind)Mo(CO)2(NCMe)2Cl] |
1954 |
1851 |
23
|
| [(η5-Ind)Mo(CO)2(NCMe)2][BF4] |
1970 |
1880 |
23
|
| [(η3-Ind)Mo(CO)2(DMF)2Cl] |
1934 |
1830 |
24
|
| [(η5-Ind)Mo(CO)2(DMF)2][BF4] |
1962 |
1865 |
24
|
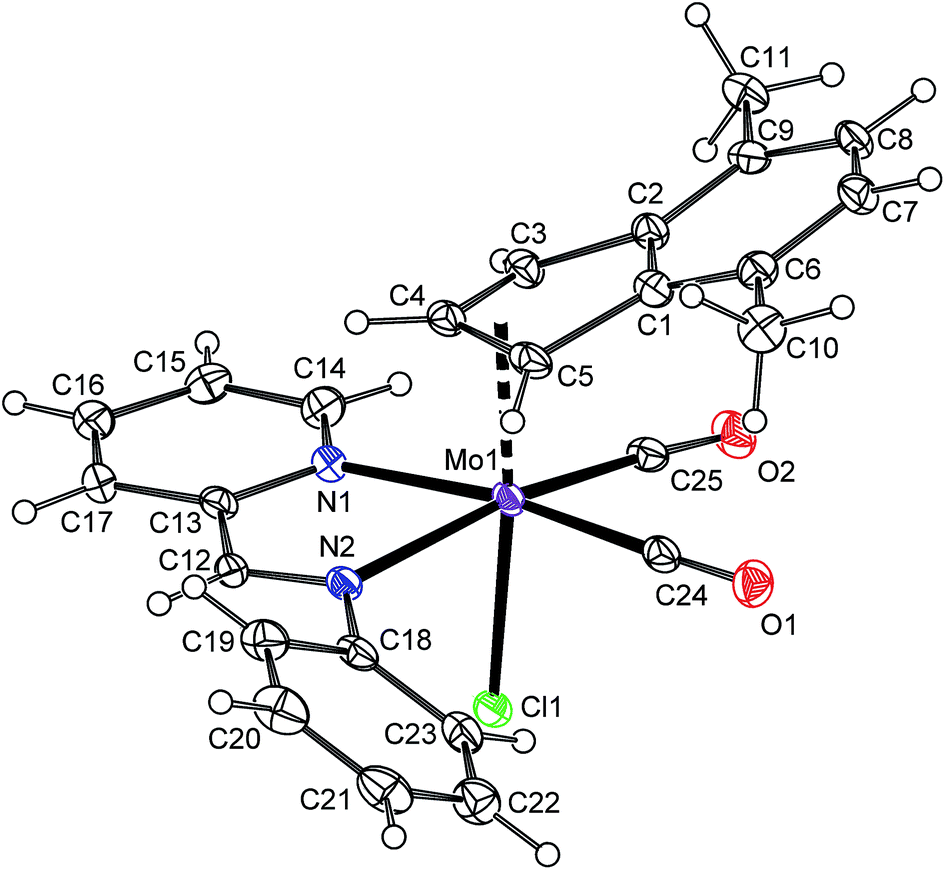
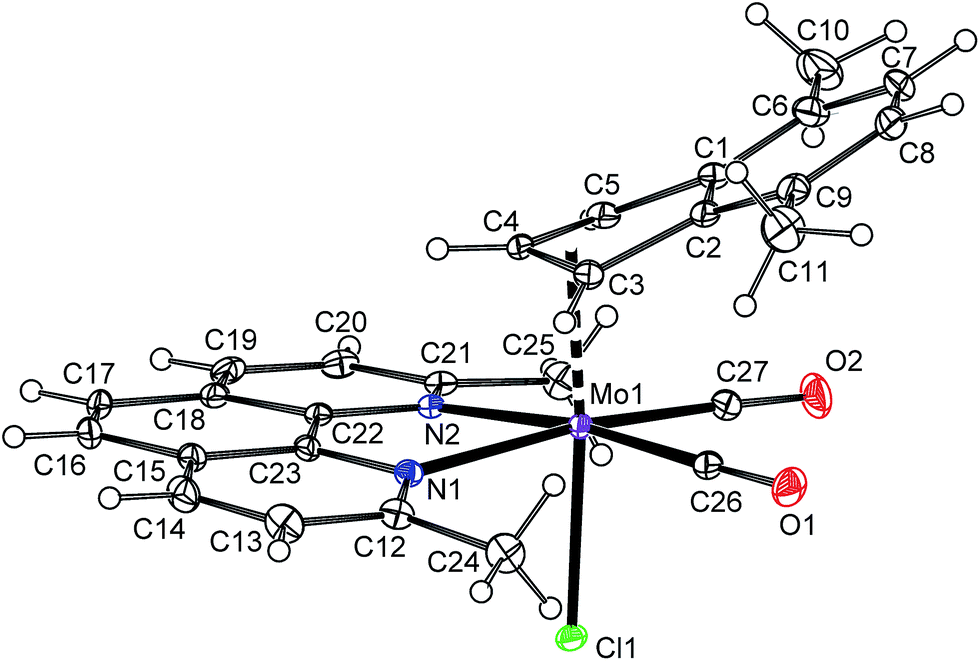
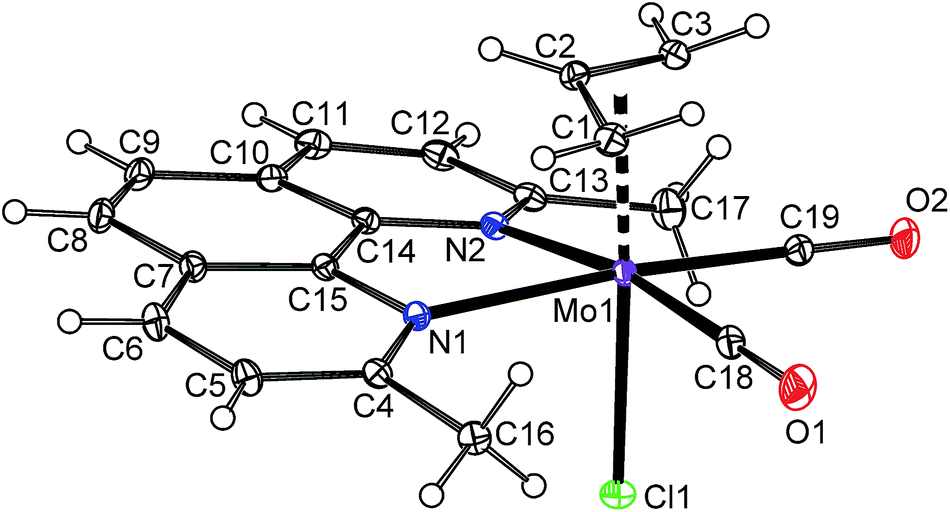
The solid state structure of the compound 6a was determined by X-ray diffraction analysis, see Fig. 2. The molecule has a distorted octahedral structure. When the bond Cg(C3)–Mo is defined as the principal axis, indenyl and chloride occupy the axial positions while the cis-coordinated carbonyl ligands and the nitrogen donor atoms of pyma are in the equatorial plane. High values of Ω [23.1(6)°] and Δ(M–C) [0.849(6) Å], observed for indenyl ligand, are in line with desired η3-coordination mode.22,42,43 According to the initial aim, the compound 6a is isostructural with allyl complex [(η3-C3H5)Mo(CO)2(pyma)Cl] (6-allyl) that was also confirmed by X-ray diffraction analysis (cf.Fig. 2 and 3). The allyl counterpart 6-allyl was prepared by a ligand exchange reaction starting from [(η3-C3H5)Mo(CO)2(NCMe)2Cl] and pyma. The geometric parameters of 6a and 6-allyl, describing the coordination sphere of molybdenum, are very similar (see Table 3). Only the distance Mo–Cg(C3) in 6a is longer due to an electron-withdrawing character of the annulated benzene ring that weakens the donor properties of indenyl.
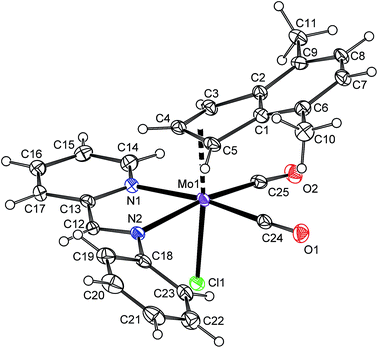 |
| | Fig. 2 ORTEP drawing of [(η3-4,7-Me2C9H5)Mo(CO)2(pyma)Cl] (6a). The labeling for all non-hydrogen atoms is shown. Thermal ellipsoids are drawn at the 30% probability level. | |
 |
| | Fig. 3 ORTEP drawing of [(η3-C3H5)Mo(CO)2(pyma)Cl] (6-allyl). The labeling for all non-hydrogen atoms is shown. Thermal ellipsoids are drawn at the 30% probability level. | |
Table 3 Geometric parameters of the neutral hexacoordinated molybdenum speciesa
| |
6a
|
6-allyl
|
10a·CH2Cl2b |
10a·CH2Cl2b |
10a·CHCl3 |
10-allyl
|
10c
|
|
Distances are given in Å; angles and dihedral angles are given in °.
Two crystallographically independent molecules in the unit cell.
For definition see footnote of Table 1.
|
| Mo–Cg(C3) |
2.1027(18) |
2.040(2) |
2.106(3) |
2.109(2) |
2.1202(16) |
2.040(5) |
2.126(3) |
| Mo–X |
2.4736(14) |
2.4957(5) |
2.4649(7) |
2.4761(7) |
2.4690(7) |
2.4858(4) |
2.6189(4) |
| Mo–C(CO) |
1.977(7) |
1.957(3) |
1.957(2) |
1.953(2) |
1.945(3) |
1.9466(14) |
1.958(3) |
| 1.964(5) |
1.959(3) |
1.958(2) |
1.957(2) |
1.966(2) |
1.9498(15) |
1.960(3) |
| Mo–N |
2.231(6) |
2.248(2) |
2.277(2) |
2.279(2) |
2.280(2) |
2.2984(11) |
2.261(2) |
| 2.262(4) |
2.279(2) |
2.283(2) |
2.281(2) |
2.287(2) |
2.2958(11) |
2.283(2) |
| Cg(C3)–Mo–X |
173.35(8) |
175.83(9) |
174.81(7) |
175.27(6) |
176.05(6) |
177.20(6) |
175.49(8) |
| C(CO)–Mo–C(CO) |
79.1(3) |
78.02(11) |
76.15(10) |
77.30(10) |
75.27(12) |
76.75(6) |
75.07(12) |
| N–Mo–N |
73.25(18) |
72.39(7) |
74.26(6) |
73.85(7) |
73.59(7) |
73.06(4) |
74.57(9) |
| Pl1–Pl2c |
2.0(3) |
0.4(1) |
10.0(2) |
14.4(2) |
13.2(2) |
14.11(8) |
9.8(2) |
|
Ω
c
|
23.1(6) |
— |
23.2(3) |
23.4(2) |
22.5(3) |
— |
21.3(3) |
| Δ(M–C)c |
0.849(6) |
— |
0.840(3) |
0.847(3) |
0.838(3) |
— |
0.805(3) |
Although the indenyl compounds 4a–6a are isostructural with the well-known allyl complexes [(η3-C3H5)Mo(CO)2(N,NL)Cl] their behavior in solution is very different. The allyl compounds are stable in solution of common organic solvents while a dissolution of the η3-indenyl compounds 4a–6a, even in non-coordinating solvents, leads to a chloride abstraction to give cationic pentacoordinated species [(η5-4,7-Me2C9H5)Mo(CO)2(N,NL)]+ (4b: N,NL = bpy, 5b: N,NL = phen, 6b: N,NL = pyma) as evidenced by the 1H NMR spectroscopy, see Scheme 3. The complexes bearing bpy (4a) and phen (5a) undergo a complete ionization in solution while the compound 6a only a partial one as evidenced by two sets of signals. It suggests that the chloride abstraction is an equilibrium reaction that is further supported by a fact that a solvent evaporation gives back the initial hexacoordinated species 4a–6a, see Scheme 3.
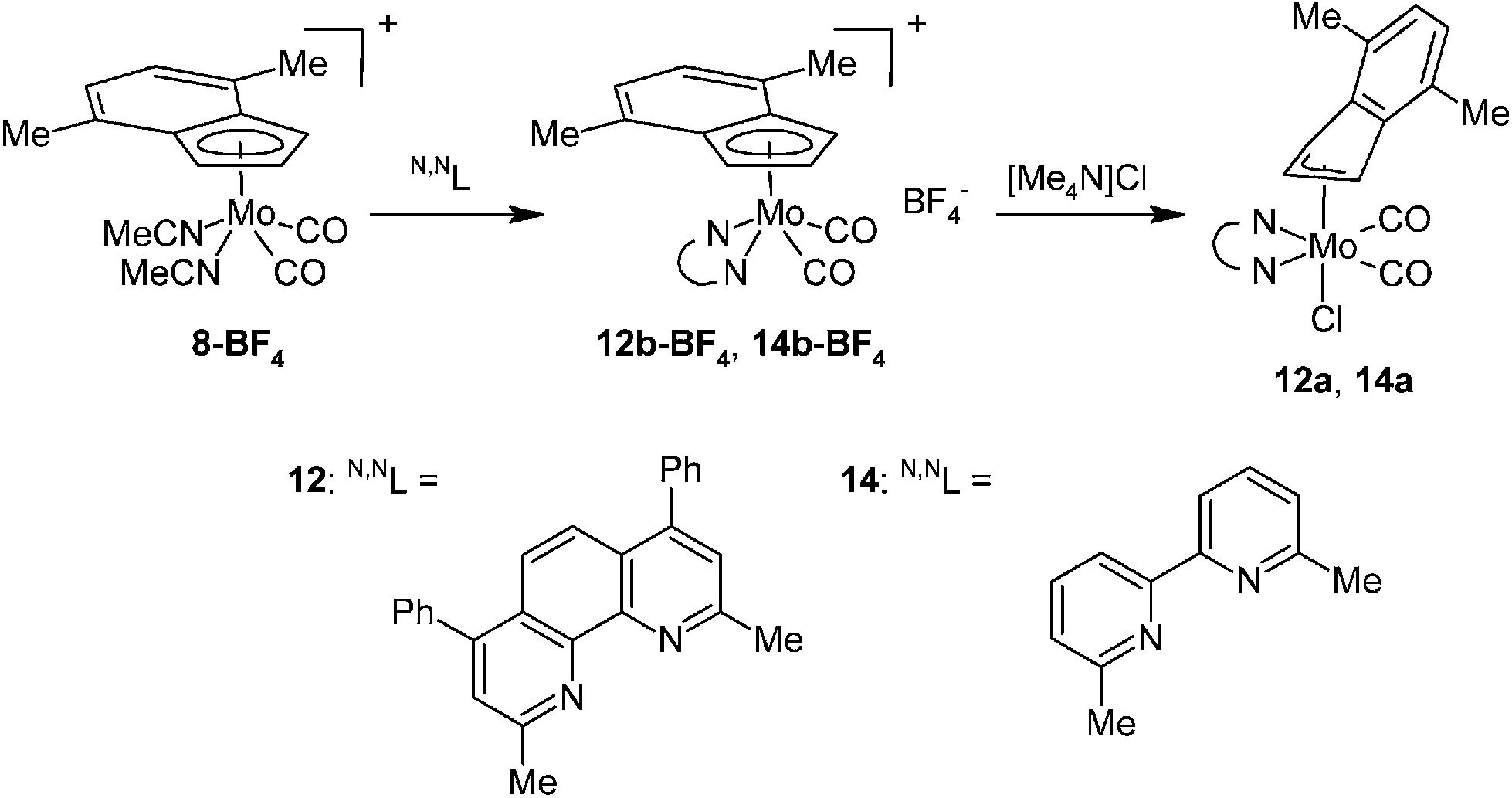
The assignment of the sets of signals to the η3 -or η5-indenyl species is done according to chemical shift of the proton at the 2-position. Hence, low-fielded triplet of H2 is diagnostic to η3-coordination mode while high-fielded one implies the η5-bonding of the indenyl ligand.40 This assignment is confirmed by the use of an alternative chloride-free protocol for the cationic species 4b and 5b compensated with a weakly coordinating tetrafluoroborate, see Scheme 4.
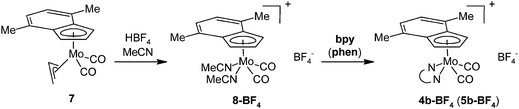 |
| | Scheme 4 Synthesis of cationic molybdenum compounds. | |
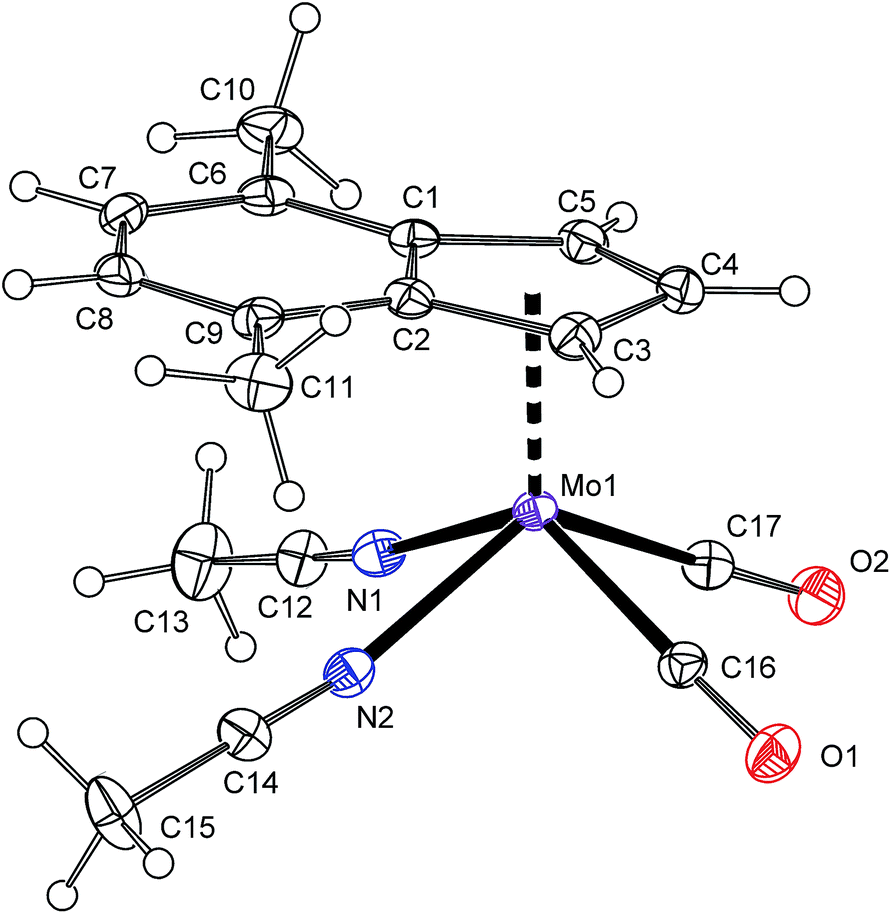
Protonation of allyl compound [(η3-C3H5)(η5-4,7-Me2C9H5)Mo(CO)2] (7) in presence of acetonitrile gives the cationic compound [(η5-4,7-Me2C9H5)Mo(CO)2(NCMe)2][BF4] (8-BF4). Following ligand exchange produces the desired chelate complexes [(η5-4,7-Me2C9H5)Mo(CO)2(bpy)][BF4] (4b-BF4) and [(η5-4,7-Me2C9H5)Mo(CO)2(phen)][BF4] (5b-BF4). As expected, the solution 1H NMR spectra are identical with chloride analogues 4b-Cl and 5b-Cl. High CO stretching frequencies observed for solid samples of 4b-BF4 and 5b-BF4 are in line with the presence of weakly coordinating anion (cf. with 4a and 5a in Table 2) and prove retention of 4b-BF4 and 5b-BF4 in the solid state. The structures of 8-BF4 and 4b-BF4 were determined by X-ray diffraction analysis. Both cations have a square-pyramidal structure, see Fig. 4 and 5. The apical position is occupied with the η5-bonded indenyl ligand (for Ω and Δ(M–C) see Table 1). The cis-coordinated carbonyl ligands and two nitrogen donor atoms of remaining ligands form the basal plane. The structural parameters, describing the coordination sphere of molybdenum, are in line with data of structurally related cyclopentadienyl and indenyl compounds described previously.13,14,44–46
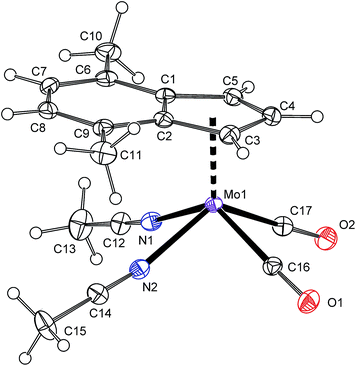 |
| | Fig. 4 ORTEP drawing of cation [(η5-4,7-Me2C9H5)Mo(CO)2(NCMe)2]+ present in the crystal structure of 8-BF4. The labeling for all non-hydrogen atoms is shown. Thermal ellipsoids are drawn at the 30% probability level. | |
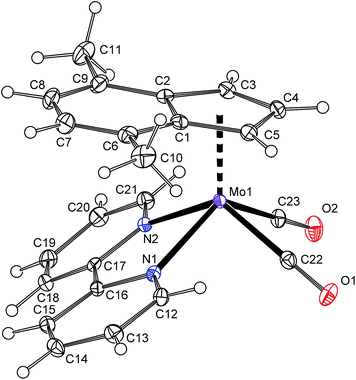 |
| | Fig. 5 ORTEP drawing of cation [(η5-4,7-Me2C9H5)Mo(CO)2(bpy)]+ present in the crystal structure of 4b-BF4. The labeling for all non-hydrogen atoms is shown. Thermal ellipsoids are drawn at the 30% probability level. | |
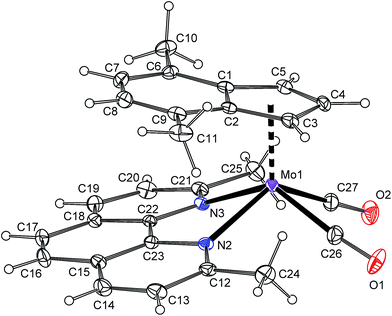
The reaction of 2 with lower than stoichiometric amount of bpy or phen gives a material insoluble in common organic solvents that strongly contrasts with properties of the starting compounds as well as the products 4a and 5a. Elemental analysis of the products together with their insolubility suggests an appearance of the ionic pair consisting of a large complex cation and a large complex anion [(η5-4,7-Me2C9H5)Mo(CO)2(N,NL)][(η5-4,7-Me2C9H5)Mo(CO)2Cl2] (4b-9: N,NL = bpy, 5b-9: N,NL = phen), see Scheme 5. Slow diffusion of a phenanthroline solution into a solution of the starting compound (2) gives single crystals suitable for the X-ray diffraction analysis that confirmed the proposed ionic structure of 5b-9 (see Fig. 6). The cationic part is isostructural with bipyridine complex 4b-BF4. The anionic part forms square-pyramid with two cis-coordinated carbonyl ligands and two chlorides in basal plane. The η5-coordinated indenyl (for Ω and Δ(M–C) see Table 4) occupies the apical position. The geometric parameters describing the coordination sphere of anionic part are very close to the data recently published for starting dimeric species [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2).22 The crystal structure of 5b-9 is stabilized by intramolecular π–π stacking involving phenanthroline ligand of 5b and indenyl ligand of 9. The distance between the centroid of central phenanthroline ring and centroid of the C6-ring of the indenyl ligand was found to be 3.493(5) Å.
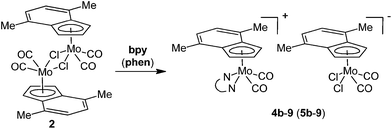 |
| | Scheme 5 Reaction of 2 with one equivalent of bpy or phen. | |
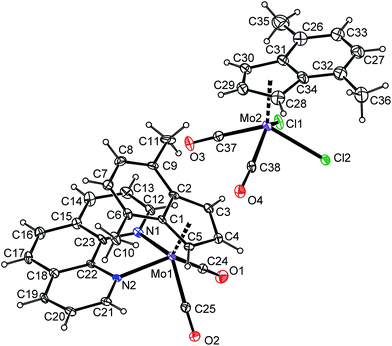 |
| | Fig. 6 ORTEP drawing of [(η5-4,7-Me2C9H5)Mo(CO)2(bpy)]+ and [(η5-4,7-Me2C9H5)Mo(CO)2Cl2]− present in the crystal structure of 5b-9·CH2Cl2. The labeling for all non-hydrogen atoms is shown. Thermal ellipsoids are drawn at the 20% probability level. | |
Table 4 Geometric parameters of the anionic molybdenum species [(η5-4,7-Me2C9H5)Mo(CO)2Cl2]− present in the crystal structure of 5b-9·CH2Cl2a
|
Distances are given in Å, angles and dihedral angles are given in °.
For definition see footnote of Table 1.
|
| Mo–Cg(C5) |
2.036(4) |
C(CO)–Mo–C(CO) |
74.0(4) |
| Mo–C(CO) |
1.942(9) |
Cl–Mo–Cl |
81.56(8) |
| 1.909(9) |
| Mo–Cl |
2.495(2) |
Ω
b
|
9.9(10) |
| 2.514(2) |
| |
|
Δ(M–C)b |
0.239(10) |
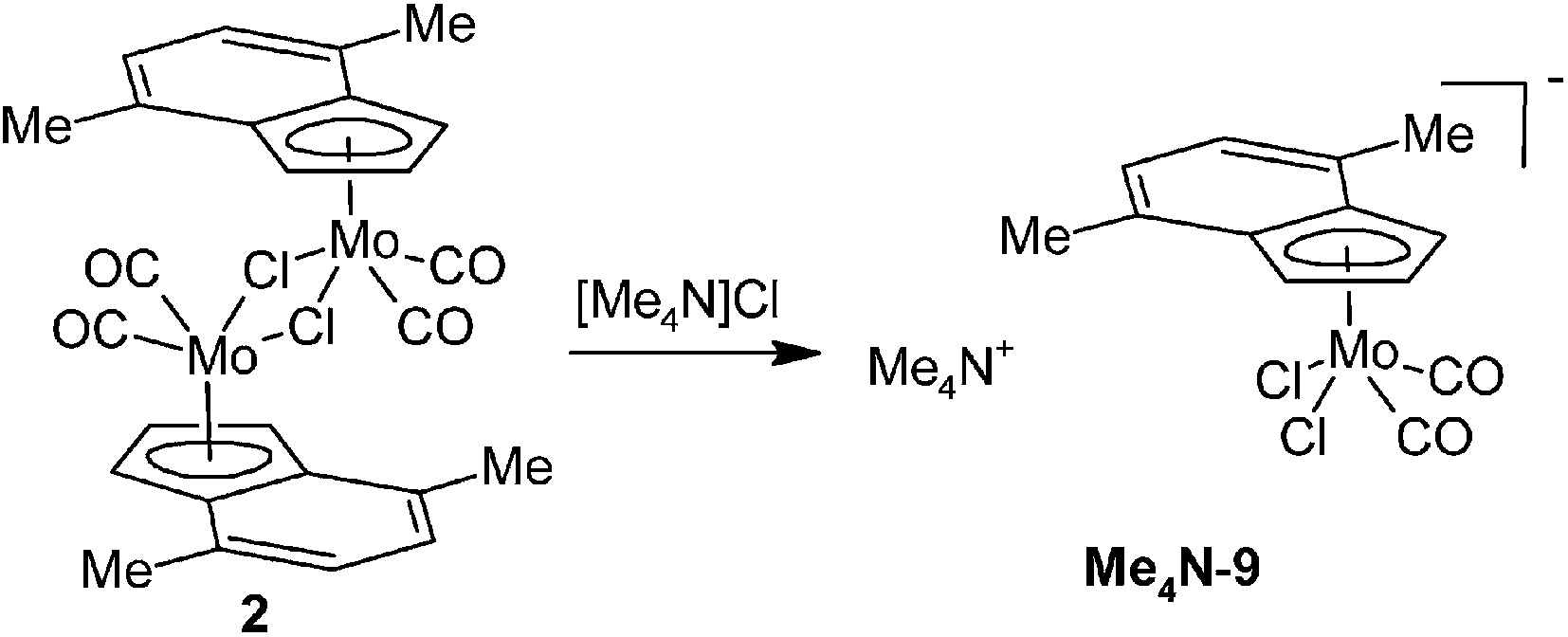
Since the anionic complex (9) is unprecedented an alternative method of the synthesis was developed. It is given by the reaction of 2 with a common source of chlorides. Namely, the soluble ammonium salt, [Me4N][(η5-4,7-Me2C9H5)Mo(CO)2Cl2] (Me4N-9), was prepared by the reaction of 2 with [Me4N]Cl (Scheme 6). The infrared spectra of Me4N-9 show the CO stretching bands at low wavenumbers [νa(CO): 1936, 1922 cm−1; νs(CO): 1820 cm−1] that is consistent with the high electron density on molybdenum. The appearance of two bands of the νa(CO) is due to a vibration coupling of the carbonyl ligands in the crystal lattice. 1H NMR spectrum of Me4N-9 shows a typical pattern of the η5-indenyl ligand with a highfielded signal of H2 (δ = 4.87 ppm).
 |
| | Scheme 6 Synthesis of [Me4N][(η5-4,7-Me2C9H5)Mo(CO)2Cl2] (Me4N-9). | |
Reaction of 2 with sterically demanding N,N-chelating ligands
Dissociation of the complexes bearing bpy, phen and pyma led us to a more sterically demanding N,N-chelating ligand, 2,9-dimethyl-1,10-phenanthroline (2,9-Me2-phen), in attempt to stabilize the η3-indenyl species in solution.
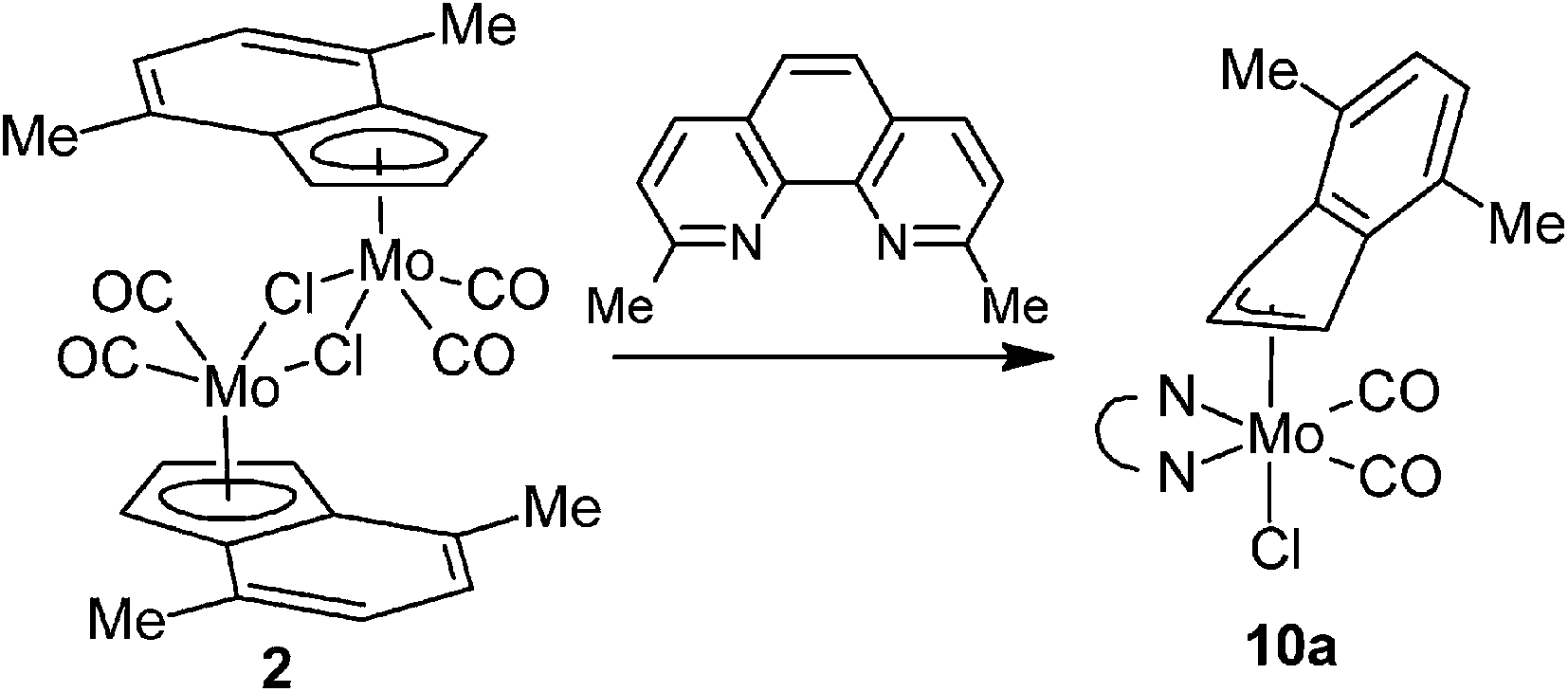
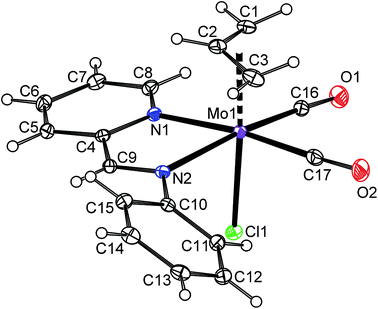
The product of the reaction between [η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2) and 2,9-Me2-phen shows similar frequencies of the CO stretching bands [νa(CO): 1927 cm−1; νs(CO): 1848 cm−1] as observed for the hexacoordinated species 4a–6a that implies a similar molecular structure in the solid state, [(η3-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)Cl] (10a), see Scheme 7. The enhanced stability of 10a in solution was confirmed by 1H NMR spectroscopy. Hence, the spectrum shows only one set of signals. The signal of the indenyl proton H2 appears at high field (δ = 5.84 ppm) that is typical for η3-coordination mode. The observed spectrum pattern is consistent with Cs molecular symmetry that confirms a configuration with the nitrogen donor atoms of the chelating ligand in trans-positions to the cis-coordinated carbonyl ligands. The X-ray structure analyses of 10a·CH2Cl2 and 10a·CHCl3 confirm appearance of this isomer in the solid state, see Fig. 7. The η3-coordination mode of the indenyl ligand is evidenced by slipping parameters Ω (∼23°) and Δ(M–C) (∼0.84 Å), see Table 3.
 |
| | Scheme 7 Synthesis of [(η3-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)Cl] (10a). | |
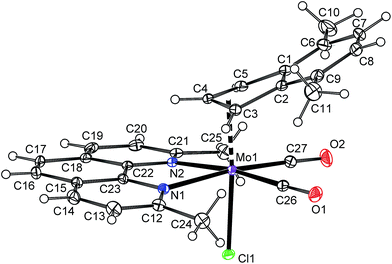 |
| | Fig. 7 ORTEP drawing of [(η3-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)Cl] present in the crystal structure of 10a·CH2Cl2. The labeling for all non-hydrogen atoms is shown. Thermal ellipsoids are drawn at the 30% probability level. Only one of two crystallographically independent molecules is shown for clarity. | |
Although the coordination sphere of molybdenum seems to be very similar to the pyma complex 6a (cf. bond distances and bond angles in Table 3), the methyl groups in the near neighborhood of carbonyl ligands prevent an appearance of a planar chelate cycle. Hence, the methyl groups of 2,9-Me2-phen are forced below the equatorial plane, defined by molybdenum and carbonyl ligands, that causes an envelope folding of the chelate cycle of ∼14°. This distortion is caused by steric repulsion of the methyl groups with the carbonyl ligands as evidenced by short nonbonding distances C(CH3)–C(CO) of 3.064(4)–3.138(4) Å those are considerably shorter than sum of van der Waals radii of two carbon atoms (3.4 Å).47 The folding of the chelate cycle seems to be independent on nature of η3-ligands. Hence, the η3-allyl analogue 10-allyl is distorted in the same way as 10a; cf.Fig. 7 and 8 and Pl1–Pl2 in Table 3.
 |
| | Fig. 8 ORTEP drawing of [(η3-C3H5)Mo(CO)2(2,9-Me2-phen)Cl] (10-allyl). The labeling for all non-hydrogen atoms is shown. Thermal ellipsoids are drawn at the 30% probability level. | |
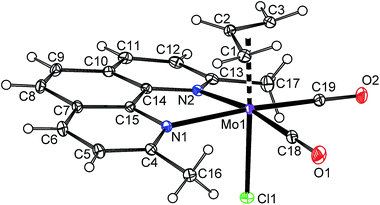
The high stability of 10a toward the loss of chloride suggests that the cationic complex bearing 2,9-Me2-phen, 10b-BF4, should be a suitable precursor for the assembly of hexacoordinated complexes bearing various anionic 2e ligands (Scheme 8). The starting 10b-BF4 was synthesized using the standard protocol outlined above for 4b-BF4. The X-ray diffraction analysis of the 10b-BF4·0.5(Me2-phen·HBF4) reveals the expected square-pyramidal structure (Fig. 9) with the η5-coordinated indenyl ligand as documented by slippage parameters [Ω = 6.3(4)°; Δ(M–C) [0.178(4) Å]. The folding of the chelate cycle [Pl1–Pl2 is 22.4(2)°] is, in this case, much heavier than observed for 2,2′-bipyridine analogue 4b-BF4 [7.8(2)°] and even that in hexacoordiated species 10a [10.0(2)–14.4(2)°]. This distortion is accompanied with prolongation of the bonds Mo–N (cf.10b-BF4·0.5(Me2-phen·HBF4) with 4b-BF4 and 5b-9·CH2Cl2 in Table 1). These observations imply that a repulsion of the phenanthroline methyl groups from the carbonyl ligands is weaker for hexacoordinated species and seems to be a driving force for its stabilization in solution. The crystal structure of 10b-BF4·0.5(Me2-phen·HBF4) reveals π–π stacking interactions involving two molecules of cationic complex 10b and a protonated ligand. The distance between centroid of the central phenanthroline rings are 3.598(3) Å.48
 |
| | Scheme 8 Optimized synthesis of η3-indenyl compounds. X = Cl, Br, I, NCO and NCS for compounds denoted a, c, d, e and f, respectively. | |
 |
| | Fig. 9 ORTEP drawing of cation [(η5-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)]+ present in the crystal structure of 10b-BF4·0.5(Me2-phen·HBF4). The labeling for all non-hydrogen atoms is shown. Thermal ellipsoids are drawn at the 20% probability level. | |
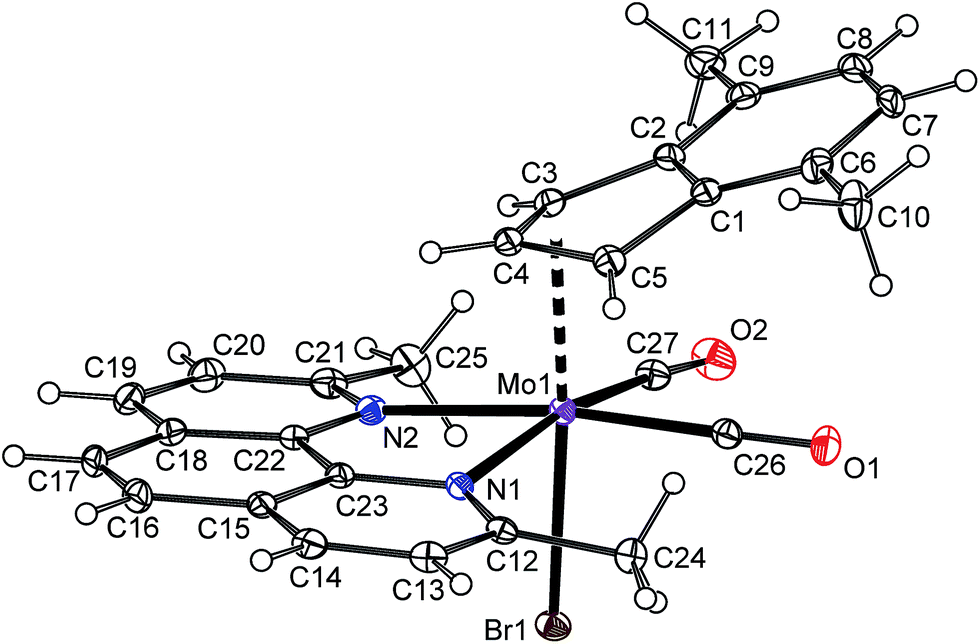
The instability of the pentacoordinated cationic species in presence of free monoanionic ligands was evidenced on reactions of 10b-BF4 with a series of halides and pseudohalides. An apearance of the hexacoordinated species [(η3-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)X] (10a: X = Cl, 10c: X = Br, 10d: X = I, 10e: X = NCO, 10f: X = NCS) is documented by the infrared and NMR spectroscopy. The compounds 10c–10f are stable in solution. The 1H NMR spectra of the isolated products confirm that the dissociation reaction is highly disfavored similarly as in case of the parent chloride complex 10a. The infrared spectra reveal that cyanate ligand in 10e and thiocyanate ligand in 10f are bonded to molybdenum via the nitrogen donor atom. It is documented by low stretching frequencies of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds (10e: 2195 cm−1, 10f: 2063 cm−1). This observation is in line with literature data reported for allyl analogues [(η3-C3H5)Mo(CO)2(Ph2PCH2CH2PPh2)(NCO)],49 [(η3-C3H5)Mo(CO)2(phen)(NCS)],50,51 [(η3-C3H4Ph)Mo(CO)2(phen)(NCS)],52 [(η3-C3H5)Mo(CO)2(NCMe)2(NCS)].53 Structure of the bromide complex 10c was determined by X-ray analysis, see Fig. 10. The coordination sphere of the central metal is very similar as in case of the chloride analogue 10a. The bond distance Mo–Br was found to be shorter [2.6189(4) Å] than in case of similar allyl complexes [(η3-C3H5)Mo(CO)2(N,NL)Br)] (N,NL = bpy: 2.650(3) Å, N,NL = phen: 2.645(2) Å] that is a result of different donor properties of the ligand in trans-possition to bromide.54
N bonds (10e: 2195 cm−1, 10f: 2063 cm−1). This observation is in line with literature data reported for allyl analogues [(η3-C3H5)Mo(CO)2(Ph2PCH2CH2PPh2)(NCO)],49 [(η3-C3H5)Mo(CO)2(phen)(NCS)],50,51 [(η3-C3H4Ph)Mo(CO)2(phen)(NCS)],52 [(η3-C3H5)Mo(CO)2(NCMe)2(NCS)].53 Structure of the bromide complex 10c was determined by X-ray analysis, see Fig. 10. The coordination sphere of the central metal is very similar as in case of the chloride analogue 10a. The bond distance Mo–Br was found to be shorter [2.6189(4) Å] than in case of similar allyl complexes [(η3-C3H5)Mo(CO)2(N,NL)Br)] (N,NL = bpy: 2.650(3) Å, N,NL = phen: 2.645(2) Å] that is a result of different donor properties of the ligand in trans-possition to bromide.54
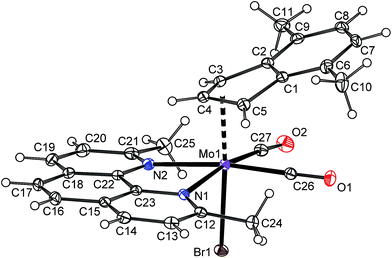 |
| | Fig. 10 ORTEP drawing of [(η3-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)Br] present in the crystal structure of 10c. The labeling for all non-hydrogen atoms is shown. Thermal ellipsoids are drawn at the 30% probability level. | |
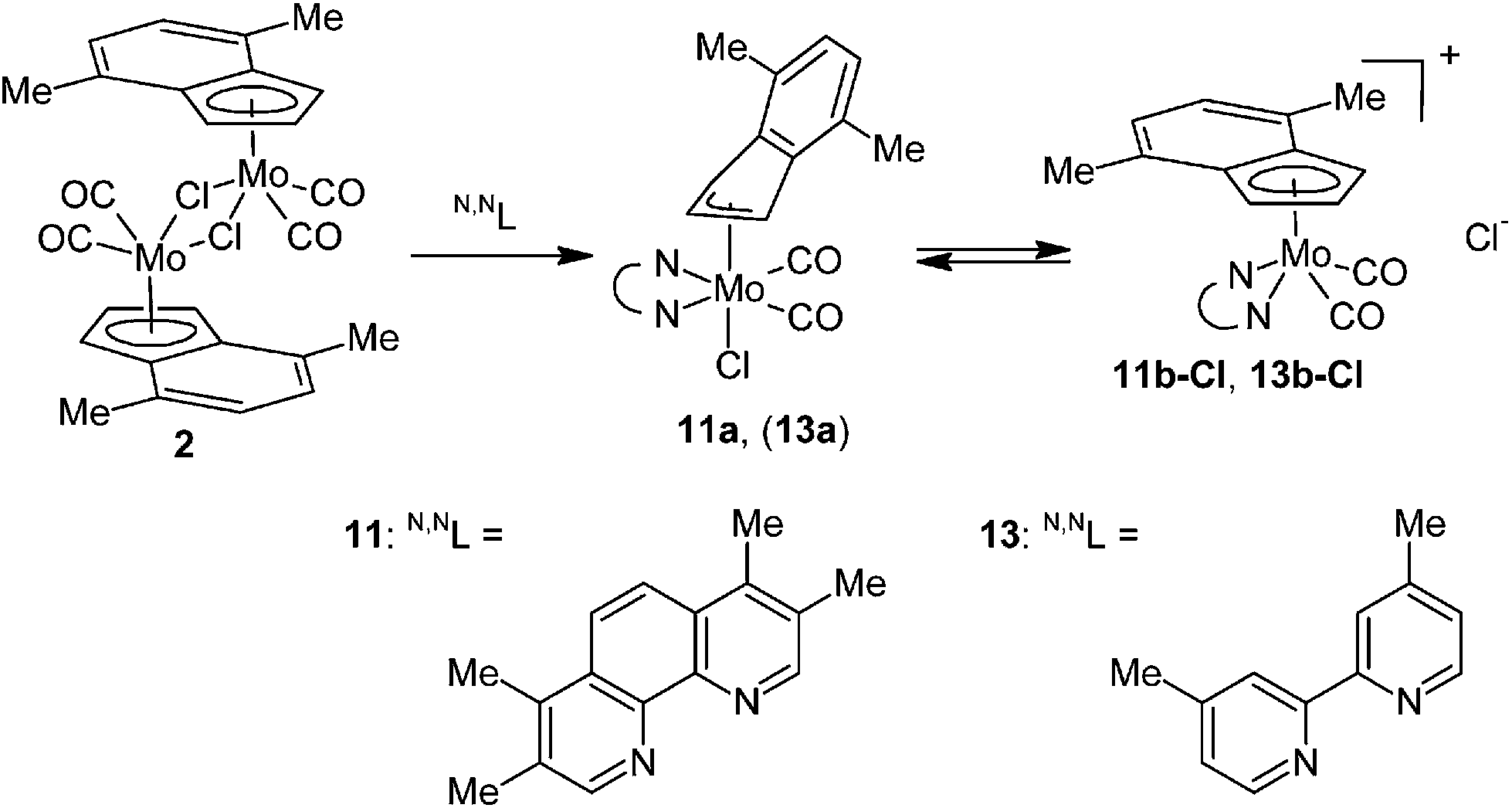
A variation of the electronic properties of the bidentate ligands has only minor effect on stability of the η3-indenyl complexes as documented on experiments with series of modified N,N-chelating ligands, namely 3,4,7,8-Me4-phen, 2,9-Me2-4,7-Ph2-phen, 4,4′-Me2-bpy and 6,6′-Me2-bpy. Although the donor ability of the 1,10-phenanthroline ligands decreases in the line 3,4,7,8-Me4-phen > 2,9-Me2-4,7-Ph2-phen > 2,9-Me2-phen > phen,55,56 the stability of the molybdenum complexes does not follow this relation. Derivatives bearing methyl groups beside the nitrogen donor atom (i.e. 2,9-Me2-phen and 2,9-Me2-4,7-Ph2-phen) form much more stable complexes (10a and 12a) than the rest of the series (i.e. phen and 3,4,7,8-Me4-phen), see Schemes 3, 9 and 10. Hence, complexes 10a and 12a are stable in solution, while compounds 5a and 11a undergo dissociation giving cationic complexes 5b-Cl and 11b-Cl, respectively. These observations reveal that the stability of the products is mainly influenced by steric effects of the chelating ligands, overshadowing the electronic ones. Similar conclusion was achieved based on experiments with dimethyl substituted 2,2′-bipyridines. Although donor ability of 4,4′-Me2-bpy is comparable to 6,6′-Me2-bpy, only the later ligand gives a stable hexacoordinated species (14a) that does not undergo dissociation in solution and cloud be easily synthesized by reaction of 14b-BF4 with common source of chlorides (e.g. [Me4N]Cl), see Scheme 10. In contrary, 4,4′-Me2-bpy produces complex with ionic structure (13b-Cl) both in solution and in solid state, see Scheme 9.
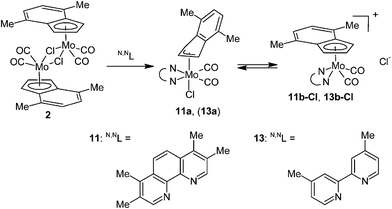 |
| | Scheme 9 Reaction of 2 with two equivalents of 3,4,7,8-Me4-phen and 4,4′-Me2-bpy. | |
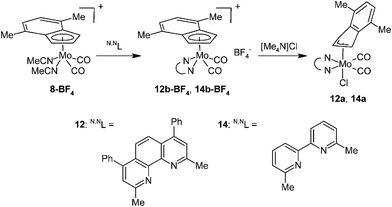 |
| | Scheme 10 Syntheses of complexes bearing 2,9-Me2-4,7-Ph2-phen and 6,6′-Me2-bpy. | |
Conclusions
Although sterically demanding ligands are commonly used for the stabilization of low-coordinate metal compounds57,58 or complexes in a low oxidation state,59,60 an application of this approach for the stabilization of low hapticity of the indenyl ligand is rather unusual.25 In current study, we clearly demonstrated that modification of the steric properties of a N,N-chelating ligand is a suitable approach for the hapticity adjustment in the indenyl molybdenum compounds. Hence, powerful steric effect of methyl groups in positions beside nitrogen donor atoms activates [(η5-4,7-Me2C9H5)Mo(CO)2]+ core toward association of 2e anionic ligand (e.g. halide) that is otherwise highly disfavored. Such nucleophile-induced indenyl ring slippage enables the assembly of η3-indenyl species isostructural with highly attended allyl complexes. This observation strongly contrasts with less demanding ligands (e.g. bpy, 4,4′-Me2-bpy, phen, 3,4,7,8-Me4-phen) those form pentacoordinated cationic species [(η5-4,7-Me2C9H5)Mo(CO)2(N,NL)]+ in solution and desired η3-compounds were stable only in particular cases in the solid state.
Experimental section
Methods and materials
All operations were performed under nitrogen using conventional Schlenk-line techniques. The solvents were purified and dried by standard methods.61 Starting materials were available commercially or prepared according to literature procedures: [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2),22 [(η3-C3H5)(η5-4,7-Me2C9H5)Mo(CO)2] (7),22 [(η3-C3H5)Mo(CO)2(NCMe)2Cl],62 The infrared spectra were recorded in the 4000−400 cm−1 region (resolution 2 cm−1) on a Nicolet Magna 6700 FTIR spectrometer using a Diamond Smart Orbit ATR. 1H and 13C{1H} NMR spectra were measured on a Bruker Avance 400 and a Bruker Avance 500 spectrometers at room temperature. The chemical shifts are given in ppm relative to TMS.
Synthesis of 6,6′-dimethyl-2,2′-bipyridine (6,6′-Me2-bpy)
A solution of bpy (1.00 g, 6.4 mmol) in THF (100 mL) was cooled at −50 °C, treated with MeLi (17 mL, 1.6 mol L−1, 27.2 mmol) and stirred at this temperature for 1 h. The solution was warmed to room temperature, stirred for another 1 h and then refluxed overnight. After cooling at room temperature, the reaction mixture was transferred into a mixture ice-water. The organic layer was separated and the aqueous layer was extracted three times with CH2Cl2. The combined organic layers were drier with MgSO4 and filtered. The volatiles were vacuum evaporated on a rotavapor. Resulting brown oil was oxidized by a saturated solution of KMnO4 in acetone until formation of MnO2 ceased. The MnO2 was removed by vacuum filtration through short pad of celite on a glass frit. Acetone was vacuum evaporated on a rotavapor and the crude product was purified by column chromatography on silica (hexane/ethyl acetate = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Rf(TLC) = 0.70 (hexane/ethyl acetate = 1:1). Yield: 0.29 g (25%, 1.57 mmol). Analytical and spectroscopic date were consistent with those reported elsewhere.63
:1). Rf(TLC) = 0.70 (hexane/ethyl acetate = 1:1). Yield: 0.29 g (25%, 1.57 mmol). Analytical and spectroscopic date were consistent with those reported elsewhere.63
Synthesis of [(η5-4,7-Me2C9H5)Mo(CO)2(py)Cl] (3)
[{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2; 200 mg, 0.30 mmol) was dissolved in CH2Cl2 (50 mL) and treated with py (475 mg, 6.0 mmol). The solution was stirred at room temperature overnight. The volatiles were vacuum evaporated. The crude product was washed with ether and recrystallized from the mixture CH2Cl2/ether and vacuum dried. Yield: 205 mg (83%, 0.50 mmol). Red powder. Mp: 150–160 °C (dec.). Anal. calcd for C18H16ClMoNO2: C, 52.77; H, 3.94; N, 3.42. Found: C, 52.98; H, 3.87; N, 3.60. 1H NMR (CDCl3; 400 MHz; δ (ppm)): 8.70 (s-br, 1H, py), 8.13 (dd, 3J(1H,1H) = 6.4 Hz, 4J(1H,1H) = 1.4 Hz, 2H, py), 7.68 (tt, 3J(1H,1H) = 7.6 Hz, 4J(1H,1H) = 1.4 Hz, 1H, py), 7.11 (m, 1H, py), 7.03 (d, 3J(1H,1H) = 7.2 Hz, 1H, C9H5, H5,6), 6.91 (d, 3J(1H,1H) = 7.2 Hz, 1H, C9H5, H5,6), 5.87 (d, 3J(1H,1H) = 2.8 Hz, 4J(1H,1H) = 1.5 Hz, 1H, C9H5, H1,3), 5.69 (d, 3J(1H,1H) = 2.8 Hz, 4J(1H,1H) = 1.5 Hz, 1H, C9H5, H1,3), 5.31 (t, 3J(1H,1H) = 2.8 Hz, 1H, C9H5, H2), 2.37 (s, 3H, CH3), 1.47 (s, 3H, CH3). FTIR (ATR, cm−1): 1940 vs [νsa(CO)], 1848 vs [νs(CO)], 1819 vs [νs(CO)]. Single crystals of 3 suitable for X-ray diffraction analysis were prepared by overlayering of the CH2Cl2 solution with hexane.
Synthesis of [(η3-4,7-Me2C9H5)Mo(CO)2(bpy)Cl] (4a)
[{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2; 200 mg, 0.30 mmol) was dissolved in CH2Cl2 (50 mL) and treated with bpy (94 mg, 0.60 mmol). The solution was stirred at room temperature overnight. The volatiles were vacuum evaporated. The crude product was washed with ether and recrystallized from the mixture CH2Cl2/ether and vacuum dried. Yield: 285 mg (98%, 0.59 mmol). Dark red powder. Mp: 150–160 °C (dec.). Anal. calcd for C23H19ClMoN2O2: C, 56.75; H, 3.93; N, 5.75. Found: C, 56.92; H, 4.12; N, 5.57. 1H NMR (CDCl3; 400 MHz; δ (ppm); 4b-Cl): 9.54 (s-br, 2H, bpy), 8.91 (d, 3J(1H,1H) = 5.5 Hz, 2H, bpy), 8.22 (s-br, 2H, bpy), 7.47 (t, 3J(1H,1H) = 6.1 Hz, 2H, bpy), 6.57 (s, 2H, C9H5, H5,6), 6.09 (d, 3J(1H,1H) = 2.6 Hz, 2H, C9H5, H1,3), 5.53 (t, 3J(1H,1H) = 2.6 Hz, 1H, C9H5, H2), 1.91 (s, 6H, CH3). FTIR (ATR, cm−1): 1940 vs [νa(CO)], 1862 vs [νs(CO)].
Synthesis of [(η5-4,7-Me2C9H5)Mo(CO)2(bpy)][BF4] (4b-BF4)
[(η5-4,7-Me2C9H5)Mo(CO)2(NCMe)2][BF4] (8-BF4; 100 mg, 0.22 mmol) was dissolved in CH2Cl2 (50 mL) and treated with bpy (34 mg, 0.22 mmol). The solution was stirred at room temperature overnight. The volatiles were vacuum evaporated. The crude product was washed with ether and recrystallized from the mixture CH2Cl2/ether and vacuum dried. Yield: 115 mg (99%, 0.21 mmol). Red powder. Mp: 180–190 °C (dec.). Anal. calcd for C23H19BF4MoN2O2: C, 51.33; H, 3.56; N, 5.21. Found: C, 51.06; H, 3.71; N, 5.18. 1H NMR (CDCl3; 400 MHz; δ (ppm)): 8.89 (d, 3J(1H,1H) = 5.7 Hz, 2H, bpy), 8.64 (d, 3J(1H,1H) = 8.6 Hz, 2H, bpy), 8.14 (t, 3J(1H,1H) = 7.8 Hz, 2H, bpy), 7.50 (t, 3J(1H,1H) = 6.5 Hz, 2H, bpy), 6.62 (s, 2H, C9H5, H5,6), 6.09 (d, 3J(1H,1H) = 2.9 Hz, 2H, C9H5, H1,3), 5.56 (t, 3J(1H,1H) = 2.9 Hz, 1H, C9H5, H2), 1.91 (s, 6H, CH3). FTIR (ATR, cm−1): 1970 vs [νa(CO)], 1894 vs [νs(CO)], 1040 vs [ν(BF)]. Single crystals of 4b-BF4 suitable for X-ray diffraction analysis were prepared by overlayering of the CH2Cl2 solution with ether.
Synthesis of [(η3-4,7-Me2C9H5)Mo(CO)2(phen)Cl] (5a)
Steps of the synthesis followed the procedure for the compounds 4a. Reagents: [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2; 200 mg, 0.30 mmol) and phen (108 mg, 0.60 mmol). Yield: 265 mg (86%, 0.52 mmol). Dark red powder. Mp: 150–160 °C (dec.). Anal. calcd for C25H19ClMoN2O2: C, 58.78; H, 3.75; N, 5.48. Found: C, 58.83; H, 3.96; N, 5.44. 1H NMR (CD3CN; 400 MHz; δ (ppm); 5b-Cl): 9.57 (dd, 3J(1H,1H) = 5.4 Hz, 4J(1H,1H) = 1.3 Hz, 2H, phen), 8.67 (dd, 3J(1H,1H) = 8.3 Hz, 4J(1H,1H) = 1.2 Hz, 2H, phen), 8.08 (s, 2H, phen), 7.94 (dd, 3J(1H,1H) = 8.2 Hz, 4J(1H,1H) = 5.2 Hz, 2H, phen), 6.45 (d, 3J(1H,1H) = 2.8 Hz, 2H, C9H5, H1,3), 6.16 (s, 2H, C9H5, H5,6), 5.69 (t, 3J(1H,1H) = 2.8 Hz, 1H, C9H5, H2), 1.84 (s, 6H, CH3). FTIR (ATR, cm−1): 1919 vs [νa(CO)], 1844 vs [νs(CO)].
Synthesis of [(η5-4,7-Me2C9H5)Mo(CO)2(phen)][BF4] (5b-BF4)
Steps of the synthesis followed the procedure for the compounds 4b-BF4. Reagents: [(η5-4,7-Me2C9H5)Mo(CO)2(NCMe)2][BF4] (8-BF4; 50 mg, 0.11 mmol) and phen (20 mg, 0.11 mmol). Yield: 65 mg (99%, 0.11 mmol). Red powder. Mp: 160–170 °C (dec.). Anal. calcd for C25H19BF4MoN2O2: C, 57.08; H, 3.14; N, 4.59. Found: C, 57.17; H, 3.19; N, 4.42. 1H NMR (CD2Cl2; 400 MHz; δ (ppm)): 9.41 (dd, 3J(1H,1H) = 5.4 Hz, 4J(1H,1H) = 1.2 Hz, 2H, phen), 8.62 (dd, 3J(1H,1H) = 8.1 Hz, 4J(1H,1H) = 1.2 Hz, 2H, phen), 8.07 (s, 2H, phen), 7.96 (dd, 3J(1H,1H) = 8.1 Hz, 3J(1H,1H) = 5.4 Hz, 2H, phen), 6.27 (s, 2H, C9H5, H5,6), 6.27 (d, 3J(1H,1H) = 2.8 Hz, 2H, C9H5, H1,3), 5.67 (t, 3J(1H,1H) = 2.8 Hz, 1H, C9H5, H2), 1.86 (s, 6H, CH3). 13C NMR (CD2Cl2; 101 MHz; δ (ppm)): 252.3 (2C, CO), 133.1, 132.3 (2 × 1Cq), 156.6, 139.0, 129.0, 128.1, 125.1 (5 × 2C, phen and C9H5), 91.3 (1C, C2 of C9H5), 79.5 (2C, C1,3 of C9H5), 19.0 (2C, CH3). FTIR (ATR, cm−1): 1967 vs [νa(CO)], 1880 vs [νs(CO)].
Synthesis of [(η3-4,7-Me2C9H5)Mo(CO)2(pyma)Cl] (6a)
Steps of the synthesis followed the procedure for the compounds 4a. Reagents: [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2; 200 mg, 0.30 mmol) and pyma (109 mg, 0.60 mmol). Yield: 285 mg (93%, 0.56 mmol). Purple powder. Mp: 150–160 °C (dec.). Anal. calcd for C25H21ClMoN2O2: C, 58.55; H, 4.13; N, 5.46. Found: C, 58.44; H, 4.25; N, 5.59. 1H NMR (CD3CN; 400 MHz; δ (ppm); 6a and 6b-Cl in the ratio 1:3): 8.61 (d, 3J(1H,1H) = 5.4 Hz, 1H of a, pyma), 8.60 (s, 1H of a, pyma), 8.41 (s, 1H of b, pyma), 8.25 (d, 3J(1H,1H) = 7.8 Hz, 1H of b, pyma), 8.10 (m, 1H of a and 1H of b, pyma), 7.60–7.25 (m, 7H of a and 7H of b, pyma), 7.20 (d, 3J(1H,1H) = 7.2 Hz, 1H of b, C9H5, H5,6), 7.07 (d, 3J(1H,1H) = 7.0 Hz, 1H of a, C9H5, H5,6), 6.93 (d, 3J(1H,1H) = 7.2 Hz, 1H of b, C9H5, H5,6), 6.64 (d, 3J(1H,1H) = 7.0 Hz, 1H of a, C9H5, H5,6), 6.61 (t, 3J(1H,1H) = 3.6 Hz, 1H of a, C9H5, H2), 6.16 (dd, 3J(1H,1H) = 2.8 Hz, 4J(1H,1H) = 1.5 Hz, 1H b, C9H5, H1,3), 6.10 (dd, 3J(1H,1H) = 2.8 Hz, 4J(1H,1H) = 1.5 Hz, 1H of b, C9H5, H1,3), 5.72 (t, 3J(1H,1H) = 2.8 Hz, 1H of b, C9H5, H2), 5.09 (dd, 3J(1H,1H) = 3.6 Hz, 4J(1H,1H) = 2.5 Hz, 1H of a, C9H5, H1,3), 4.25 (dd, 3J(1H,1H) = 3.6 Hz, 4J(1H,1H) = 2.5 Hz, 1H of a, C9H5, H1,3), 2.07 (s, 3H of a, CH3), 2.06 (s, 3H of b, CH3), 1.73 (s, 3H of a, CH3), 1.57 (s, 3H of b, CH3). FTIR (ATR, cm−1): 1934 vs [νa(CO)], 1855 vs [νs(CO)]. Single crystals of 6a suitable for X-ray diffraction analysis were prepared by overlayering of the CHCl3 solution with hexane.
Synthesis of [(η3-C3H5)Mo(CO)2(pyma)Cl] (6-allyl)
[(η3-C3H5)Mo(CO)2(NCMe)2Cl] (200 mg, 0.64 mmol) was dissolved in CH2Cl2 (50 mL) and treated with pyma (117 mg, 0.64 mmol). The solution was stirred at room temperature overnight. The volatiles were vacuum evaporated. The crude product was washed with ether and recrystallized from the mixture CH2Cl2/ether and vacuum dried. Yield: 250 mg (95%, 0.61 mmol). Dark puple powder. Mp: 160–170 °C (dec.). Anal. calcd for C17H15ClMoN2O2: C, 49.72; H, 3.68; N, 6.82. Found: C, 49.61; H, 3.70; N, 6.94. 1H NMR (CD3CN; 400 MHz; δ (ppm): 8.84 (d, 3J(1H,1H) = 5.1 Hz, 1H, pyma), 8.53 (s, 1H, pyma), 8.10 (td, 3J(1H,1H) = 7.8 Hz, 4J(1H,1H) = 1.4 Hz 1H, pyma), 7.98 (d, 3J(1H,1H) = 7.6 Hz, 1H, pyma), 7.65–7.40 (m, 6H, pyma), 3.36 (tt, 3J(1H,1H) = 9.2 Hz, 3J(1H,1H) = 6.2 Hz, 1H, meso-C3H5), 3.16 (dd, 3J(1H,1H) = 6.2 Hz, 4J(1H,1H) = 3.3 Hz, 1H, syn-C3H5), 2.48 (dd, 3J(1H,1H) = 6.2 Hz, 4J(1H,1H) = 3.3 Hz, 1H, syn-C3H5), 1.31 (d, 3J(1H,1H) = 9.2 Hz, 1H, anti-C3H5), 1.02 (d, 3J(1H,1H) = 9.2 Hz, 1H, anti-C3H5). FTIR (ATR, cm−1): 1929 vs [νa(CO)], 1845 vs [νs(CO)]. Single crystals of 6-allyl suitable for X-ray diffraction analysis were prepared by overlayering of the CH2Cl2 solution with hexane.
Synthesis of [(η5-4,7-Me2C9H5)Mo(CO)2(NCMe)2][BF4] (8-BF4)
[(η3-C3H5)(η5-4,7-Me2C9H5)Mo(CO)2] (7; 500 mg, 1.49 mmol) was dissolved in CH2Cl2 (50 mL), cooled at 0 °C and treated with acetonitrile (5 mL) and then with HBF4·Et2O (1.49 mmol). The reaction mixture was stirred for one hour at room temperature. Afterwards, the solvent was vacuum evaporated. The crude product was washed with ether and recrystallized from the mixture CH2Cl2/ether and vacuum dried. Yield: 608 mg (88%, 1.31 mmol). Orange powder. Mp: 150–160 °C (dec.). Anal. calcd for C17H17BF4MoN2O2: C, 44.00; H, 3.69; N, 6.04. Found: C, 43.88; H, 3.73; N, 6.12. 1H NMR (CD2Cl2; 400 MHz; δ (ppm)): 7.23 (s, 2H, C9H5, H5,6), 6.00 (d, 3J(1H,1H) = 2.6 Hz, 2H, C9H5, H1,3), 5.01 (t, 3J(1H,1H) = 2.6 Hz, 1H, C9H5, H2), 2.36 (s, 6H, CH3CN), 1.97 (s, 6H, CH3). FTIR (ATR, cm−1): 1970 vs [νa(CO)], 1897 vs [νs(CO)], 1040 vs [ν(BF)]. Single crystals of 8-BF4 suitable for X-ray diffraction analysis were prepared by overlayering of the MeCN solution with Et2O.
Synthesis of [(η5-4,7-Me2C9H5)Mo(CO)2(bpy)][(η5-4,7-Me2C9H5)Mo(CO)2Cl2] (4b-9)
[{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2; 100 mg, 0.15 mmol) was dissolved in CH2Cl2 (50 mL) and treated with bpy (23 mg, 0.15 mmol). The solution was stirred at room temperature overnight. The precipitate was decanted. The product was washed with ether, CH2Cl2 and vacuum dried. Yield: 85 mg (69%, 0.10 mmol). Red powder. Mp: 140–150 °C (dec.). Anal. calcd for C36H30Cl2Mo2N2O4: C, 52.90; H, 3.70; N, 3.43. Found: C, 52.82; H, 3.79; N, 3.51. FTIR (ATR, cm−1): 1950 vs [νa(CO)], 1930 vs [νa(CO)], 1875 vs [νs(CO)], 1837 vs [νs(CO)].
Synthesis of [(η5-4,7-Me2C9H5)Mo(CO)2(phen)][(η5-4,7-Me2C9H5)Mo(CO)2Cl2] (5b-9)
Steps of the synthesis followed the procedure for the compounds 4b-9. Reagents: [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2; 100 mg, 0.15 mmol) and phen (27 mg, 0.15 mmol). Yield: 95 mg (75%, 0.11 mmol). Red powder. Mp: 140–150 °C (dec.). Anal. calcd for C38H30Cl2Mo2N2O4: C, 54.24; H, 3.59; N, 3.33. Found: C, 54.38; H, 3.62; N, 3.10. FTIR (ATR, cm−1): 1954 vs [νa(CO)], 1926 vs [νa(CO)], 1867 vs [νs(CO)], 1824 vs [νs(CO)]. Single crystals of 5b-9·CH2Cl2 suitable for X-ray diffraction analysis were prepared by overlayering of the CH2Cl2 solutions of the starting materials.
Synthesis of [Me4N][(η5-4,7-Me2C9H5)Mo(CO)2Cl2] (Me4N-9)
[{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2; 100 mg, 0.15 mmol) was dissolved in acetone (50 mL) and treated with [Me4N]Cl (82 mg, 0.75 mmol). The solution was stirred at room temperature overnight. The volatiles were vacuum evaporated. The residuum was dissolved in CH2Cl2 and filtered over short pad of Celite. The volatiles were vacuum evaporated. The crude product was washed with ether and recrystallized from the mixture CH2Cl2/ether and vacuum dried. Yield: 220 mg (83%, 0.50 mmol). Red powder. Mp: 140–150 °C (dec.). Anal. calcd for C17H23Cl2MoNO2: C, 46.38; H, 5.27; N, 3.18. Found: C, 46.12; H, 5.32; N, 3.23. 1H NMR (CDCl3; 400 MHz; δ (ppm)): 6.90 (s, 2H, C9H5, H5,6), 5.76 (d, 3J(1H,1H) = 2.8 Hz, 2H, C9H5, H1,3), 4.87 (t, 3J(1H,1H) = 2.8 Hz, 1H, C9H5, H2), 3.32 (s, 12H, (CH3)4N)), 2.29 (s, 6H, (CH3)2C9H5)). FTIR (ATR, cm−1): 1936 vs [νa(CO)], 1922 vs [νa(CO)], 1820 vs [νs(CO)].
Synthesis of [(η3-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)Cl] (10a)
Method A: steps of the synthesis followed the procedure for the compounds 4a. Reagents: [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2; 200 mg, 0.30 mmol) and 2,9-Me2-phen (125 mg, 0.60 mmol). Yield: 315 mg (97%, 0.58 mmol). Method B: [(η5-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)][BF4] (10b-BF4; 100 mg, 0.20 mmol) was dissolved in acetone (50 mL) and treated with [Me4N]Cl (220 mg, 2.01 mmol; alternatively 2.01 mmol of NaCl or KCl could be used). The solution was stirred for 48 h. The volatiles were vacuum evaporated. The residuum was dissolved in CH2Cl2 and filtered over short pad of Celite. The volatiles were vacuum evaporated. The crude product was washed with ether and recrystallized from the mixture CH2Cl2/ether and vacuum dried. Yield: 105 mg (97%, 0.19 mmol). Dark red powder. Mp: 150–160 °C (dec.). Anal. calcd for C27H23ClMoN2O2: C, 60.18; H, 4.30; N, 5.19. Found: C, 59.96; H, 4.42; N, 5.27. 1H NMR (CDCl3; 400 MHz; δ (ppm)): 8.30 (d, 3J(1H,1H) = 8.2 Hz, 2H, C12H6N2), 7.88 (s, 2H, C12H6N2), 7.65 (d, 3J(1H,1H) = 8.2 Hz, 2H, C12H6N2), 6.21 (s, 2H, C9H5, H5,6), 5.84 (t, 3J(1H,1H) = 3.7 Hz, 1H, C9H5, H2), 4.57 (d, 3J(1H,1H) = 3.7 Hz, 2H, C9H5, H1,3), 3.07 (s, 6H, (CH3)2C12H6N2), 2.09 (s, 6H, (CH3)2C9H5). 13C NMR (CD2Cl2; 126 MHz; δ (ppm)): 231.0 (2C, CO), 162.3, 146.1, 145.9, 129.6, 125.7 (5 × 1Cq), 138.9, 127.1, 126.7, 125.7 (4 × 2C C12H6N2 and C9H5), 103.5 (1C, C2 of C9H5), 69.5 (2C, C1,3 of C9H5), 28.9 (2C, C12H6N2(CH3)2), 17.8 (2C, C9H5(CH3)2). FTIR (ATR, cm−1): 1927 vs [νa(CO)], 1848 vs [νs(CO)]. Single crystals of 10a·CH2Cl2 suitable for X-ray diffraction analysis were prepared by overlayering of the CH2Cl2 solution with hexane. Single crystals of 10a·CHCl3 suitable for X-ray diffraction analysis were prepared by overlayering of the CHCl3 solution with ether.
Synthesis of [(η3-C3H5)Mo(CO)2(2,9-Me2-phen)Cl] (10-allyl)
Steps of the synthesis followed the procedure for the compounds 6-allyl. Reagents: [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (200 mg, 0.64 mmol) and 2,9-Me2-phen (135 mg, 0.64 mmol). Yield: 260 mg (93%, 0.60 mmol). Dark orange powder. Mp: 150–160 °C (dec.). Anal. calcd for C19H17ClMoN2O2: C, 52.25; H, 3.92; N, 6.41. Found: C, 52.38; H, 3.86; N, 6.49. 1H NMR (acetone-d6; 400 MHz; δ (ppm)): 8.60 (d, 3J(1H,1H) = 8.3 Hz, 2H, C12H6N2), 8.07 (s, 2H, C12H6N2), 7.93 (d, 3J(1H,1H) = 8.3 Hz, 2H, C12H6N2), 3.27 (s, 6H, CH3), 2.69 (d, 3J(1H,1H) = 6.2 Hz, 2H, syn-C3H5), 2.63 (tt, 1H, meso-C3H5), 1.13 (d, 3J(1H,1H) = 9.0 Hz, 2H, anti-C3H5). FTIR (ATR, cm−1): 1929 vs [νa(CO)], 1836 vs [νs(CO)]. Single crystals of 10-allyl suitable for X-ray diffraction analysis were prepared by overlayering of the CH2Cl2 solution with hexane.
Synthesis of [(η5-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)][BF4] (10b-BF4)
Steps of the synthesis followed the procedure for the compounds 4b-BF4. Reagents: [(η5-4,7-Me2C9H5)Mo(CO)2(NCMe)2][BF4] (8-BF4; 500 mg, 1.08 mmol) and 2,9-Me2-phen (224 mg, 1.08 mmol). Yield: 505 mg (96%, 1.03 mmol). Red powder. Mp: 160–170 °C (dec.). Anal. calcd for C19H17BF4MoN2O2: C, 46.75; H, 3.51; N, 5.74. Found: C, 46.92; H, 3.35; N, 5.78. 1H NMR (CD2Cl2; 400 MHz; δ (ppm)): 8.50 (d, 3J(1H,1H) = 8.3 Hz, 2H, C12H6N2), 7.96 (s, 2H, C12H6N2), 7.86 (d, 3J(1H,1H) = 8.3 Hz, 2H, C12H6N2), 6.27 (d, 3J(1H,1H) = 2.9 Hz, 2H, C9H5, H1,3), 6.04 (s, 2H, C9H5, H5,6), 5.45 (t, 3J(1H,1H) = 2.9 Hz, 1H, C9H5, H2), 3.60 (s, 6H, (CH3)2C12H6N2), 1.74 (s, 6H, (CH3)2C9H5). 13C NMR (CD2Cl2; 101 MHz; δ (ppm)): 252.5 (2C, CO), 166.5, 146.2, 132.9, 131.5, 121.0 (5 × 1Cq), 140.2, 130.0, 127.3, 127.3 (4 × 2C C12H6N2 and C9H5), 90.6 (1C, C2 of C9H5), 79.9 (2C, C1,3 of C9H5), 32.8 (2C, C12H6N2(CH3)2), 18.6 (2C, C9H5(CH3)2). FTIR (ATR, cm−1): 1946 vs [νa(CO)], 1867 vs [νs(CO)]. Single crystals of 10b-BF4·0.5(Me2-phen·HBF4) suitable for X-ray diffraction analysis were prepared by overlayering of the crude product in CH2Cl2 with hexane.
Synthesis of [(η3-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)Br] (10c)
Steps of the synthesis followed the procedure for the compounds 10a (Method B). Reagents: [(η5-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)][BF4] (10b-BF4; 100 mg, 0.20 mmol) and KBr (240 mg, 2.02 mmol). Yield: 90 mg (94%, 0.19 mmol). Dark red powder. Mp: 160–170 °C (dec.). Anal. calcd for C19H17BrMoN2O2: C, 47.42; H, 3.56; N, 5.82. Found: C, 47.30; H, 3.62; N, 5.95. 1H NMR (CDCl3; 400 MHz; δ (ppm)): 8.30 (d, 3J(1H,1H) = 8.3 Hz, 2H, C12H6N2), 7.89 (s, 2H, C12H6N2), 7.65 (d, 3J(1H,1H) = 8.3 Hz, 2H, C12H6N2), 6.21 (s, 2H, C9H5, H5,6), 5.83 (t, 3J(1H,1H) = 3.7 Hz, 1H, C9H5, H2), 4.61 (d, 3J(1H,1H) = 3.7 Hz, 2H, C9H5, H1,3), 3.04 (s, 6H, (CH3)2C12H6N2), 2.08 (s, 6H, (CH3)2C9H5). 13C NMR (CD2Cl2; 101 MHz; δ (ppm)): 230.1 (2C, CO), 162.6, 146.2, 145.9, 129.7, 125.8 (5 × 1Cq), 138.8, 127.1, 126.6, 125.9 (4 × 2C C12H6N2 and C9H5), 103.6 (1C, C2 of C9H5), 68.6 (2C, C1,3 of C9H5), 30.1 (2C, C12H6N2(CH3)2), 17.8 (2C, C9H5(CH3)2). FTIR (ATR, cm−1): 1936 vs [νa(CO)], 1863 vs [νs(CO)]. Single crystals of 10c suitable for X-ray diffraction analysis were prepared by overlayering of the CH2Cl2 solution with Et2O.
Synthesis of [(η3-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)I] (10d)
Steps of the synthesis followed the procedure for the compounds 10a (Method B). Reagents: [(η5-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)][BF4] (10b-BF4; 100 mg, 0.20 mmol) and KI (330 mg, 1.99 mmol). Yield: 100 mg (95%, 0.19 mmol). Brown powder. Mp: 160–170 °C (dec.). Anal. calcd for C19H17IMoN2O2: C, 43.20; H, 3.24; N, 5.30. Found: C, 43.44; H, 3.34; N, 5.43. 1H NMR (CDCl3; 400 MHz; δ (ppm)): 8.29 (d, 3J(1H,1H) = 8.2 Hz, 2H, C12H6N2), 7.91 (s, 2H, C12H6N2), 7.65 (d, 3J(1H,1H) = 8.2 Hz, 2H, C12H6N2), 6.21 (s, 2H, C9H5, H5,6), 5.84 (s-br, 1H, C9H5, H2), 4.69 (d, 3J(1H,1H) = 3.4 Hz, 2H, C9H5, H1,3), 3.00 (s, 6H, (CH3)2C12H6N2), 2.07 (s, 6H, (CH3)2C9H5). 13C NMR (CD2Cl2; 126 MHz; δ (ppm)): 229.0 (2C, CO), 162.9, 146.3, 145.8, 129.8, 125.9 (5 × 1Cq), 138.8, 127.2, 126.5, 125.9 (4 × 2C C12H6N2 and C9H5), 104.3 (1C, C2 of C9H5), 67.5 (2C, C1,3 of C9H5), 30.5 (2C, C12H6N2(CH3)2), 17.9 (2C, C9H5(CH3)2). FTIR (ATR, cm−1): 1932 vs [νa(CO)], 1861 vs [νs(CO)].
Synthesis of [(η3-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)(NCO)] (10e)
Steps of the synthesis followed the procedure for the compounds 10a (Method B). Reagents: [(η5-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)][BF4] (10b-BF4; 100 mg, 0.20 mmol) and KOCN (160 mg, 1.97 mmol). Yield: 80 mg (90%, 0.18 mmol). Dark red powder. Mp: 160–170 °C (dec.). Anal. calcd for C20H17MoN3O3: C, 54.19; H, 3.87; N, 9.48. Found: C, 54.01; H, 3.89; N, 9.32. 1H NMR (CDCl3; 400 MHz; δ (ppm)): 8.34 (d, 3J(1H,1H) = 8.3 Hz, 2H, C12H6N2), 7.94 (s, 2H, C12H6N2), 7.71 (d, 3J(1H,1H) = 8.3 Hz, 2H, C12H6N2), 6.20 (s, 2H, C9H5, H5,6), 5.88 (t, 3J(1H,1H) = 3.7 Hz, 1H, C9H5, H2), 4.68 (d, 3J(1H,1H) = 3.7 Hz, 2H, C9H5, H1,3), 3.02 (s, 6H, (CH3)2C12H6N2), 2.08 (s, 6H, (CH3)2C9H5). FTIR (ATR, cm−1): 2195 vs [ν(CN)], 1930 vs [νa(CO)], 1851 vs [νs(CO)].
Synthesis of [(η3-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)(NCS)] (10f)
Steps of the synthesis followed the procedure for the compounds 10a (Method B). Reagents: [(η5-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-phen)][BF4] (10b-BF4; 100 mg, 0.20 mmol) and KSCN (200 mg, 2.06 mmol). Yield: 90 mg (98%, 0.19 mmol). Red powder. Mp: 150–160 °C (dec.). Anal. calcd for C20H17MoN3O2S: C, 52.29; H, 3.73; N, 9.15, S, 6.98. Found: C, 52.09; H, 3.65; N, 9.00, S, 7.12. 1H NMR (CDCl3; 400 MHz; δ (ppm)): 8.42 (d, 3J(1H,1H) = 8.3 Hz, 2H, C12H6N2), 7.96 (s, 2H, C12H6N2), 7.73 (d, 3J(1H,1H) = 8.3 Hz, 2H, C12H6N2), 6.21 (s, 2H, C9H5, H5,6), 5.79 (t, 3J(1H,1H) = 3.7 Hz, 1H, C9H5, H2), 4.77 (d, 3J(1H,1H) = 3.7 Hz, 2H, C9H5, H1,3), 3.04 (s, 6H, (CH3)2C12H6N2), 2.08 (s, 6H, (CH3)2C9H5). FTIR (ATR, cm−1): 2063 vs [ν(CN)], 1925 vs [νa(CO)], 1849 vs [νs(CO)].
Synthesis of [(η3-4,7-Me2C9H5)Mo(CO)2(3,4,7,8-Me4-phen)Cl] (11a)
Steps of the synthesis followed the procedure for the compounds 4a. Reagents: [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2; 50 mg, 76 μmol) and 3,4,7,8-Me4-phen (35 mg, 0.15 mmol). Yield: 50 mg (93%, 88 μmol). Red powder. Mp: 140–150 °C (dec.). Anal. calcd for C25H21ClMoN2O2: C, 61.44; H, 4.80; N, 5.64. Found: C, 61.28; H, 4.87; N, 5.55. 1H NMR (CD2Cl2; 400 MHz; δ (ppm); 11a and 11b-Cl in the ratio 1:3): 9.13 (s, 2H of b, C12H4N2), 8.59 (s, 2H of a, C12H4N2), 8.17 (s, 2H of b, C12H4N2), 8.14 (s, 2H of a, C12H4N2), 6.32 (d, 3J(1H,1H) = 2.8 Hz, 2H of b, C9H5, H1,3), 6.28 (s, 2H of b, C9H5, H5,6), 6.22 (t, 3J(1H,1H) = 3.7 Hz, 1H of a, C9H5, H2), 6.21 (s, 2H of a, C9H5, H5,6), 5.70 (t, 3J(1H,1H) = 2.8 Hz, 1H of b, C9H5, H2), 4.93 (d, 3J(1H,1H) = 3.7 Hz, 2H of a, C9H5, H1,3), 2.82 (s, 6H of b, (CH3)4C12H4N2), 2.76 (s, 6H of a, (CH3)4C12H4N2), 2.66 (s, 6H of b, (CH3)4C12H4N2), 2.58 (s, 6H of a, (CH3)4C12H4N2), 2.16 (s, 6H of a, (CH3)2C9H5), 1.88 (s, 6H of b, (CH3)2C9H5). FTIR (ATR, cm−1): 1922 vs [νa(CO)], 1839 vs [νs(CO)].
Synthesis of [(η3-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-4,7-Ph2-phen)Cl] (12a)
Steps of the synthesis followed the procedure for the compounds 10a (Method B). Reagents: [(η5-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-4,7-Ph2-phen)][BF4] (12b-BF4; 30 mg, 40 μmol) and [Me4N]Cl (44 mg, 0.40 mmol). Yield: 27 mg (97%, 39 μmol). Purple powder. Mp: 170–180 °C (dec.). Anal. calcd for C39H31ClMoN2O2:C, 67.78; H, 4.52; N, 4.05. Found: C, 67.89; H, 4.47; N, 4.19. 1H NMR (CD2Cl2; 500 MHz; δ (ppm)): 7.94 (s, 2H, C12H4N2), 7.66 (s, 2H, C12H4N2), 7.62–7.55 (m, 10H, C6H5), 6.21 (s, 2H, C9H5, H5,6), 6.07 (t, 3J(1H,1H) = 3.7 Hz, 1H, C9H5, H2), 4.70 (d, 3J(1H,1H) = 3.7 Hz, 2H, C9H5, H1,3), 3.07 (s, 6H, (CH3)2C12H4N2), 2.10 (s, 6H, (CH3)2C9H5). 13C NMR (CD2Cl2; 126 MHz; δ (ppm)): 231.1 (2C, CO), 161.5, 151.2, 146.7, 146.1, 136.9, 127.8, 125.7 (7 × 1Cq), 130.1, 129.4 (2 × 4C C6H5), 129.8, 127.1, 125.7, 124.8 (4 × 2C C12H4N2, C6H5 and C9H5), 103.8 (1C, C2 of C9H5), 69.4 (2C, C1,3 of C9H5), 30.1 (2C, C12H4N2(CH3)2), 17.8 (2C, C9H5(CH3)2). FTIR (ATR, cm−1): 1939 vs [νa(CO)], 1862 vs [νs(CO)].
Synthesis of [(η5-4,7-Me2C9H5)Mo(CO)2(2,9-Me2-4,7-Ph2-phen)][BF4] (12b-BF4)
Steps of the synthesis followed the procedure for the compounds 4b-BF4. Reagents: [(η5-4,7-Me2C9H5)Mo(CO)2(NCMe)2][BF4] (8-BF4; 50 mg, 0.11 mmol) and 2,9-Me2-4,7-Ph2-phen (36 mg, 0.11 mmol). Yield: 78 mg (98%, 0.11 mmol). Red powder. Mp: 150–160 °C (dec.). Anal. Calcd for C39H31BF4MoN2O2:C, 63.09; H, 4.21; N, 3.77. Found: C, 63.25; H, 4.14; N, 3.89. 1H NMR (CD2Cl2; 500 MHz; δ (ppm)): 7.94 (s, 2H, C12H4N2), 7.82 (s, 2H, C12H4N2), 7.66–7.52 (m, 10H, C6H5), 4.31 (d, 3J(1H,1H) = 2.8 Hz, 2H, C9H5, H1,3), 6.11 (s, 2H, C9H5, H5,6), 5.49 (t, 3J(1H,1H) = 2.8 Hz, 1H, C9H5, H2), 3.63 (s, 6H, (CH3)2C12H4N2), 1.79 (s, 6H, (CH3)2C9H5). 13C NMR (CD2Cl2; 126 MHz; δ (ppm)): 252.4 (2C, CO), 166.0, 152.1, 147.1, 135.7, 133.0, 121.3 (6 × 1Cq), 130.0, 129.8 (2 × 4C C6H5), 130.7, 129.8, 127.6, 124.9 (4 × 2C C12H4N2, C6H5 and C9H5), 91.1 (1C, C2 of C9H5), 79.8 (2C, C1,3 of C9H5), 33.1 (2C, C12H4N2(CH3)2), 18.8 (2C, C9H5(CH3)2). FTIR (ATR, cm−1): 1944 vs [νa(CO)], 1893 s [νs(CO)], 1877 s [νs(CO)].
Synthesis of [(η5-4,7-Me2C9H5)Mo(CO)2(4,4′-Me2-bpy)][Cl] (13b-Cl)
Steps of the synthesis followed the procedure for the compounds 4a. Reagents: [{(η5-4,7-Me2C9H5)Mo(CO)2(μ-Cl)}2] (2; 50 mg, 76 μmol) and 4,4′-Me2-bpy (28 mg, 0.15 mmol). Yield: 75 mg (96%, 0.15 mmol). Red powder. Mp: 130–140 °C (dec.). Anal. calcd for C25H23ClMoN2O2: C, 58.32; H, 4.50; N, 5.44. Found: C, 58.45; H, 4.61; N, 5.52. 1H NMR (CD2Cl2; 400 MHz; δ (ppm)): 8.84 (d, 3J(1H,1H) = 5.4 Hz, 2H, C10H6N2), 8.72 (s, 2H, C10H6N2), 7.32 (d, 3J(1H,1H) = 5.4 Hz, 2H, C10H6N2), 6.60 (s, 2H, C9H5, H5,6), 6.16 (s-br, C9H5, H1,3), 5.55 (s-br, 1H, C9H5, H2), 2.62 (s, 6H, (CH3)2C10H6N2), 1.96 (s, 6H, (CH3)2C9H5). FTIR (ATR, cm−1): 1962 vs [νa(CO)], 1877 vs [νs(CO)].
Synthesis of [(η3-4,7-Me2C9H5)Mo(CO)2(6,6′-Me2-bpy)Cl] (14a)
Steps of the synthesis followed the procedure for the compounds 10a (Method B). Reagents: [(η5-4,7-Me2C9H5)Mo(CO)2(6,6′-Me2-bpy)][BF4] (14b-BF4; 30 mg, 53 μmol) and [Me4N]Cl (58 mg, 0.53 mmol). Yield: 27 mg (99%, 52 μmol). Purple powder. Mp: 170–180 °C (dec.). Anal. calcd for C25H23ClMoN2O2: C, 58.32; H, 4.50; N, 5.44. Found: C, 58.39; H, 4.56; N, 5.45. 1H NMR (CD2Cl2; 400 MHz; δ (ppm)): 8.09 (d, 3J(1H,1H) = 7.8 Hz, 2H, C10H6N2), 7.91 (dd, 3J(1H,1H) = 7.8 Hz, 3J(1H,1H) = 7.6 Hz, 2H, C10H6N2), 7.40 (d, 3J(1H,1H) = 7.6 Hz, 2H, C10H6N2), 6.18 (s, 2H, C9H5, H5,6), 6.09 (t, 3J(1H,1H) = 3.7 Hz, 1H, C9H5, H2), 4.55 (d, 3J(1H,1H) = 3.7 Hz, 2H, C9H5, H1,3), 2.79 (s, 6H, (CH3)2C10H6N2), 2.05 (s, 6H, (CH3)2C9H5). FTIR (ATR, cm−1): 1930 vs [νa(CO)], 1852 vs [νs(CO)].
Synthesis of [(η5-4,7-Me2C9H5)Mo(CO)2(6,6′-Me2-bpy)][BF4] (14b-BF4)
Steps of the synthesis followed the procedure for the compounds 4b-BF4. Reagents: [(η5-4,7-Me2C9H5)Mo(CO)2(NCMe)2][BF4] (8-BF4; 50 mg, 0.11 mmol) and 6,6′-Me2-bpy (20 mg, 0.11 mmol). Yield: 60 mg (98%, 0.11 mmol). Red powder. Mp: 160–170 °C (dec.). Anal. calcd for C25H23BF4MoN2O2: C, 53.03; H, 4.09; N, 5.95. Found: C, 53.10; H, 4.12; N, 5.92. 1H NMR (CD2Cl2; 400 MHz; δ (ppm)): 8.15 (d, 3J(1H,1H) = 8.0 Hz, 2H, C10H6N2), 7.99 (dd, 3J(1H,1H) = 8.0 Hz, 3J(1H,1H) = 7.6 Hz, 2H, C10H6N2), 7.52 (dd, 3J(1H,1H) = 7.6 Hz, 4J(1H,1H) = 1.0 Hz, 2H, C10H6N2), 6.55 (s, 2H, C9H5, H5,6), 6.19 (d, 3J(1H,1H) = 2.9 Hz, 2H, C9H5, H1,3), 5.31 (t, 3J(1H,1H) = 2.9 Hz, 1H, C9H5, H2), 3.35 (s, 6H, (CH3)2C10H6N2), 1.84 (s, 6H, (CH3)2C9H5). FTIR (ATR, cm−1): 1947 vs [νa(CO)], 1870 vs [νs(CO)].
X-ray crystallography
The X-ray data for the crystals of the compounds 3, 4b-BF4, 5b-9·CH2Cl2, 6a, 6-allyl, 8-BF4, 10a·CH2Cl2, 10a·CHCl3, 10-allyl, 10b-BF4·0.5(phen·HBF4) and 10c and were obtained at 150 K using an Oxford Cryostream low-temperature device on a Nonius KappaCCD diffractometer with Mo Kα radiation (λ = 0.71073 Å) and a graphite monochromator. Data reductions were performed with DENZO-SMN.64 The absorption was corrected by integration methods65 and only in cases of 5b-9·CH2Cl2 and 6a it was performed analytically using SADABS software.66 Structures were solved by direct methods (Sir92)67 and refined by full-matrix least squares based on F2 (SHELXL).68 Thermal ellipsoids of C13 and C14 atoms in 5b-9·CH2Cl2, of F1, F3 and F4 atoms in 8-BF4 and of F1 atom in 10b-BF4·0.5(phen·HBF4) were treated with standard ISOR instruction implemented in SHELXL97 software.68 Hydrogen atoms were mostly localized on a difference Fourier map. However, to ensure uniformity of the treatment of the crystal, all hydrogen atoms were recalculated into idealized positions (riding model) and assigned temperature factors Uiso(H) = 1.2[Ueq(pivot atom)] or 1.5Ueq for the methyl moiety with C–H = 0.96, 0.97, and 0.93 Å for methyl, methylene, and hydrogen atoms in aromatic rings or the allyl moiety, respectively.
Acknowledgements
This work was supported by Ministry of Education of the Czech Republic (Project no. SG350004).
References
- E. Hevia, J. Pérez, L. Riera, V. Riera, I. del Río, S. García-Granda and D. Miguel, Chem.–Eur. J., 2002, 8, 4510–4521 CrossRef CAS
 .
.
- L. Coue, L. Cuesta, D. Morales, J. A. Halfen, J. Pérez, L. Riera, V. Riera, D. Miguel, N. G. Connelly and S. Boonyuen, Chem.–Eur. J., 2004, 10, 1906–1912 CrossRef CAS PubMed .
- J. Pérez, L. Riera, V. Riera, S. García-Granda, E. García-Rodríguez and D. Miguel, Chem. Commun., 2002, 384–385 RSC .
- D. Morales, J. Pérez, L. Riera, V. Riera, D. Miguel, M. E. G. Mosquera and S. García-Granda, Chem.–Eur. J., 2003, 9, 4132–4143 CrossRef CAS PubMed .
- D. Morales, M. E. N. Clemente, J. Pérez, L. Riera, V. Riera and D. Miguel, Organometallics, 2003, 22, 4124–4128 CrossRef CAS .
- E. Hevia, J. Pérez, L. Riera, V. Riera and D. Miguel, Organometallics, 2002, 21, 1750–1752 CrossRef CAS .
- J. Pérez, L. Riera, V. Riera, S. García-Granda, E. García-Rodríguez and D. Miguel, Organometallics, 2002, 21, 1622–1626 CrossRef .
- W. Chen, K. Sana, Y. Jiang, E. V. S. Meyer, S. Lapp, M. R. Galinski and L. S. Liebeskind, Organometallics, 2013, 32, 7594–7611 CrossRef CAS .
- T. C. Coombs, W. Huang, E. C. Garnier-Amblard and L. S. Liebeskind, Organometallics, 2010, 29, 5083–5097 CrossRef CAS PubMed .
- J. C. Alonso, P. Neves, M. J. P. da Silva, S. Quintal, P. D. Vaz, C. Silva, A. A. Valente, P. Ferreira, M. J. Calhorda, V. Félix and M. G. B. Drew, Organometallics, 2007, 26, 5548–5556 CrossRef CAS .
- C. A. Gamelas, A. C. Gomes, S. M. Bruno, F. A. A. Paz, A. A. Valente, M. Pillinger, C. C. Romão and I. S. Gonçalves, Dalton Trans., 2012, 3474–3484 RSC .
-
M. R. P. Norton de Matos, C. C. Romão, C. C. L. Pereira, S. S. Rodrigues, M. Mora, M. J. P. Silva, P. M. Alves and C. A. Reis, International Patent WO/2005/087783, 2005 .
- J. Honzíček, J. Vinklárek, Z. Padělková, L. Šebestová, K. Foltánová and M. Řezáčová, J. Organomet. Chem., 2012, 716, 258–268 CrossRef PubMed .
- J. Honzíček, J. Vinklárek, M. Erben, Z. Padělková, L. Šebestová and M. Řezáčová, J. Organomet. Chem., 2014, 749, 387–393 CrossRef PubMed .
- M. Abrantes, A. M. Santos, J. Mink, F. E. Kühn and C. C. Romão, Organometallics, 2003, 22, 2112–2118 CrossRef CAS .
- S. Huber, M. Cokoja and F. E. Kühn, J. Organomet. Chem., 2014, 751, 25–32 CrossRef CAS PubMed .
- M. Abrantes, S. Gago, A. A. Valente, M. Pillinger, I. S. Gonçalves, T. M. Santos, J. Rocha and C. C. Romão, Eur. J. Inorg. Chem., 2004, 4914–4920 CrossRef CAS PubMed .
- A. Capapé, A. Raith, E. Herdtweck, M. Cokoja and F. E. Kühn, Adv. Synth. Catal., 2010, 352, 547–556 CrossRef PubMed .
- S. Li, C. W. Kee, K. W. Huang, T. S. A. Hor and J. Zhao, Organometallics, 2010, 29, 1924–1933 CrossRef CAS .
- C. A. Gamelas, T. Lourenço, A. P. da Costa, A. L. Simplício, B. Royo and C. C. Romão, Tetrahedron Lett., 2008, 49, 4708–4712 CrossRef CAS PubMed .
- L. F. Veiros, C. A. Gamelas, M. J. Calhorda and C. C. Romão, Organometallics, 2011, 30, 1454–1465 CrossRef CAS .
- J. Honzíček, J. Vinklárek, M. Erben, J. Lodinský, L. Dostál and Z. Padělková, Organometallics, 2013, 32, 3502–3511 CrossRef .
- S. M. Bruno, A. C. Gomes, C. A. Gamelas, M. Abrantes, M. C. Oliveira, A. A. Valente, F. A. A. Paz, M. Pillinger, C. C. Romão and I. S. Gonçalves, New J. Chem., 2014, 38, 3172–3180 RSC .
- A. C. Gomes, C. A. Gamelas, J. A. Fernandes, F. A. A. Paz, P. Nunes, M. Pillinger, I. S. Gonçalves, C. C. Romão and M. Abrantes, Eur. J. Inorg. Chem., 2014, 3681–3689 CrossRef CAS PubMed .
- J. M. O'Connor and C. P. Casey, Chem. Rev., 1987, 87, 307–318 CrossRef .
- H. Bang, T. J. Lynch and F. Basolo, Organometallics, 1992, 11, 40–48 CrossRef CAS .
- M. E. Rerek, L. N. Ji and F. Basolo, J. Chem. Soc., Chem. Commun., 1983, 1208–1209 RSC .
- C. P. Casey and J. M. O'Connor, Organometallics, 1985, 4, 384–388 CrossRef CAS .
- J. S. Merola, R. T. Kacmarcik and D. Van Engent, J. Am. Chem. Soc., 1986, 108, 329–331 CrossRef CAS .
- C. C. Romão, Appl. Organomet. Chem., 2000, 14, 539–548 CrossRef .
- M. J. Calhorda, C. C. Romão and L. F. Veiros, Chem.–Eur. J., 2002, 8, 868–875 CrossRef CAS .
- M. J. Calhorda and L. F. Veiros, Coord. Chem. Rev., 1999, 185–186, 37–51 CrossRef CAS .
- C. A. Gamelas, N. A. G. Bandeira, C. C. L. Pereira, M. J. Calhorda, E. Herdtweck, M. Machuqueiro, C. C. Romão and L. F. Veiros, Dalton Trans., 2011, 10513–10525 RSC .
- J. Honzíček, F. A. A. Paz and C. C. Romão, Eur. J. Inorg. Chem., 2007, 2827–2838 CrossRef PubMed .
- J. Honzíček, C. C. Romão, M. J. Calhorda, A. Mukhopadhyay, J. Vinklárek and Z. Padělková, Organometallics, 2011, 30, 717–725 CrossRef .
- O. J. Curnow and G. M. Fern, J. Organomet. Chem., 2005, 690, 3018–3026 CrossRef CAS PubMed .
- M. B. Meredith, J. A. Crisp, E. D. Brady, T. P. Hanusa, G. T. Yee, M. Pink, W. W. Brennessel and V. G. Young, Organometallics, 2008, 27, 5464–5473 CrossRef CAS .
- R. B. King and K. N. Chen, Inorg. Chem., 1977, 16, 3372–3376 CrossRef CAS .
- J. Honzíček, A. Mukhopadhyay and C. C. Romão, Inorg. Chim. Acta, 2010, 363, 1601–1603 CrossRef PubMed .
- M. J. Calhorda, C. A. Gamelas, I. S. Gonçalves, E. Herdtweck, C. C. Romão and L. F. Veiros, Organometallics, 1998, 17, 2597–2611 CrossRef CAS .
- J. W. Faller, R. H. Crabtree and A. Habib, Organometallics, 1985, 4, 929–935 CrossRef CAS .
- J. Honzíček, I. Honzíčková, J. Vinklárek and Z. Růžičková, J. Organomet. Chem., 2014, 772–773, 299–306 CrossRef PubMed .
- C. A. Gamelas, E. Herdtweck, J. P. Lopes and C. C. Romão, Organometallics, 1999, 18, 506–515 CrossRef CAS .
- C. C. L. Pereira, P. J. Costa, M. J. Calhorda, C. Freire, S. S. Rodrigues, E. Herdtweck and C. C. Romão, Organometallics, 2006, 25, 5223–5234 CrossRef CAS .
- J. Honzíček, A. Mukhopadhyay, T. S. Silva, M. J. Romão and C. C. Romão, Organometallics, 2009, 28, 2871–2879 CrossRef .
- J. Schejbal, J. Honzíček, J. Vinklárek, M. Erben and Z. Růžičková, Eur. J. Inorg. Chem., 2014, 5895–5907 CrossRef CAS PubMed .
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS .
- One referee suggested that this π–π stacking may be responsible for folding of the chelate cycle instead of the described repulsion forces. Since it is difficult to separate these two factors, it is subject of our ongoing deeper investigation.
- J. W. Faller, D. A. Haitko, R. D. Adams and D. F. Chodosh, J. Am. Chem. Soc., 1979, 101, 865–876 CrossRef CAS .
- C. G. Hull and M. H. B. Stiddard, J. Organomet. Chem., 1967, 9, 519–525 CrossRef CAS .
- A. J. Graham and R. H. Fenn, J. Organomet. Chem., 1970, 25, 173–191 CrossRef CAS .
- N. W. Murrall and A. J. Welch, J. Organomet. Chem., 1986, 301, 109–130 CrossRef CAS .
- W. Goodyear, C. W. Hemingway, R. W. Harrington, M. R. Wiseman and B. J. Brisdon, J. Organomet. Chem., 2002, 664, 176–181 CrossRef .
- J. R. Ascenso, C. G. de Azevedo, M. J. Calhorda, M. A. A. F. d. C. T. Carrondo, P. Costa, A. R. Dias, M. G. B. Drew, V. Félix, A. M. Galvão and C. C. Romão, J. Organomet. Chem., 2001, 632, 197–208 CrossRef CAS .
- G. G. Siegel and T. Zeegers-Huyskens, Spectrochim. Acta, Part A, 1989, 45, 1297–1304 CrossRef .
- C. Crotti, E. Farnetti, S. Filipuzzi, M. Stener, E. Zangrando and P. Moras, Dalton Trans., 2007, 133–142 RSC .
- D. J. Mindiola, Acc. Chem. Res., 2006, 39, 813–821 CrossRef CAS PubMed .
- P. L. Holland, Acc. Chem. Res., 2008, 41, 905–914 CrossRef CAS PubMed .
- P. P. Power, J. Organomet. Chem., 2004, 689, 3904–3919 CrossRef CAS PubMed .
- P. J. Chirik, Organometallics, 2010, 29, 1500–1517 CrossRef CAS .
-
W. L. F. Armarego and D. D. Perrin, in Purification of Laboratory Chemicals, Oxford, 1996 Search PubMed .
- J. W. Faller, C. C. Chen, M. J. Mattina and A. Jakubowski, J. Organomet. Chem., 1973, 52, 361–386 CrossRef CAS .
- L. Y. Liao, X. R. Kong and X. F. Duan, J. Org. Chem., 2014, 79, 777–782 CrossRef CAS PubMed .
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS .
-
P. Coppens, in Crystallographic Computing, ed. F. R. Ahmed, S. R. Hall and C. P. Huber, Copenhagen, 1970 Search PubMed .
-
G. M. Sheldrick, in SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2002 Search PubMed .
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435–436 Search PubMed .
-
G. M. Sheldrick, in SHELXL97, University of Göttingen, Germany, 2008 Search PubMed .
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence