
Global structure of C2B4H4: hypercloso or not
Abstract
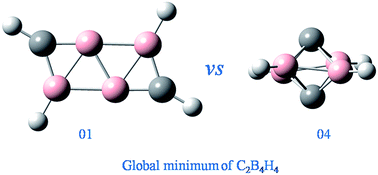
Due to their fantastic structures and reactivity, carbon–boron mixed cluster hydrides have aroused increasing attention both experimentally and theoretically. Generally, n-vertex borane and carborane containing equal to or more than (n + 1) polyhedral skeletal electron pairs (PSEPs) present similar structural characteristics. However, for the n-PSEP boranes and carboranes, whether their structural pattern is similar or not is still uncertain. In this work, we report a particular example, C2B4H4, the global minimum of which does not conform to the well-known polyhedral skeletal electron pair theory (PSEPT). Through extensive isomeric searching, the global minimum structure of C2B4H4 is addressed to exhibit a ribbon-like structure at the aug-cc-pVTZ-CCSD(T)//B3LYP level. However, the hypercloso structure predicted by PSEPT lies 16.0 kcal mol−1 higher. Our results are also different from a recent theoretical study. These findings are believed to enrich the understanding of hypercloso chemistry of carbon-substituted borane derivatives.
 Please wait while we load your content...
Please wait while we load your content...

