
Synthesis of ferrocene-containing six-membered cyclic ureas via α-ferrocenyl carbocations†
Abstract
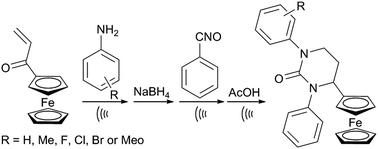
A series of ferrocene-containing six-membered cyclic ureas (1-aryl-4-ferrocenyl-3-phenyltetrahydropyrimidin-2(1H)-ones) was synthesized (in high-to-excellent yields) by reacting the corresponding aminopropanols with phenyl isocyanate and the subsequent intramolecular cyclization of the thus obtained β-hydroxy ureas (prompted by acetic acid), via an α-ferrocenyl carbocation.
 Please wait while we load your content...
Please wait while we load your content...

