
Graphitic carbon nitride (g-C3N4) as a metal-free catalyst for thermal decomposition of ammonium perchlorate†
Qi Lia,
Yi He*b and
Rufang Peng*a
aState Key Laboratory Cultivation Base for Non-metal Composites and Functional Materials, Southwest University of Science and Technology, Mianyang 621010, P. R. China. E-mail: pengrufang@swust.edu.cn
bCollege of Defence Technology, Southwest University of Science and Technology, Mianyang, 621010, P. R. China. E-mail: yhe2014@126.com
First published on 17th February 2015
Abstract
Development of metal-free and environmentally friendly catalysts is of great significance for thermal decomposition of ammonium perchlorate (AP). In the present study, graphitic carbon nitride (g-C3N4) has been demonstrated to possess intrinsic catalytic activity for thermal decomposition of AP. Adding 10 wt% g-C3N4 to AP decreases the decomposition temperature by 70 °C and the activation energy (Ea) by 119.8 kJ mol−1. Moreover, the apparent decomposition heat of AP in the presence of 10 wt% g-C3N4 reaches up to 1362.6 J g−1, which is much higher than that of the pure AP (574.2 J g−1). Furthermore, a possible catalytic mechanism for the thermal decomposition of AP with g-C3N4 has been proposed. The unique surface structure of g-C3N4 consisting of triazine units connected by planar amino groups can be favourable for the adsorption and diffusion of perchloric acid via Lewis acid–base interactions, which decreases the reaction activation energy of AP and facilitates the formation of superoxide radical anions (˙O2−) and holes (h+), leading to the oxidation reaction of ammonia more completely in the catalytic decomposition of AP.
Introduction
Ammonium perchlorate (AP), as the most common oxidizer, plays a key role in composite solid rocket propellant, which generally accounts for 60–90% of the total mass of the propellant.1 The thermal decomposition process of AP directly influences the combustion process of the propellant. A lower high-temperature thermal decomposition (HTD) temperature and reaction activation energy usually cause a shorter ignition delay time, and a higher burning rate.2 It is widely accepted that decreasing on-set temperature of HTD and increasing decomposition heat of AP are important to improve the performance of propellants. Therefore, development of excellent catalysts is an urgent requirement for the decomposition of AP. To date, various catalysts have been reported for AP thermal decomposition, especially transition metal oxides, for example, Mn3O4,3,4 CoO,5,6 MgAl2O4,7 MnO2 (ref. 8) and Al(OH)3·Cr(OH)3 (ref. 9) have been proved to be quite effective for thermal decomposition of AP. However, these catalysts suffer from many inevitable disadvantages, such as the high toxicity of heavy metal ions, high cost, and complicated preparation process, which results in environmental pollution and greatly limits their applications in modern military and industrial fields. Therefore, development of metal-free and environmentally friendly catalysts for AP thermal decomposition is highly desirable.Recently, two dimension graphitic carbon nitride (g-C3N4) as a novel polymeric semiconductor has received significant attention owing to its high nitrogen content as well as excellent chemical and thermal stability.10–15 Additionally, g-C3N4 is low cost and environmentally friendly because it can be easily prepared by condensation of urea, dicyandiamide or thiourea at elevated temperatures and does not contain any metal elements.16–18 It has been found several applications in photo catalytic fields, sensing, and bio-imaging.19–22 Nevertheless, the application of g-C3N4 as a catalyst for AP thermal decomposition has not yet been reported.
In the present study, for the first time we discovered that g-C3N4 can be a novel metal-free and environmentally friendly catalyst for thermal decomposition of AP. It can effectively decrease the activation energy of AP decomposition from 216.0 kJ mol−1 to 122.6 kJ mol−1. Moreover, the HTD temperature of AP was reduced by 70 °C and the heat release was increased by 788.4 J g−1 in the presence of 10 wt% g-C3N4. Furthermore, a possible mechanism of the thermal decomposition of AP with g-C3N4 has been discussed. The unique surface structure of g-C3N4 consisting of triazine units connected by planar amino groups can be favourable for the adsorption and diffusion of perchloric acid via Lewis acid–base interactions, which decreases the reaction activation energy of AP and facilitates the formation of superoxide radical anions and holes, leading to the oxidation reaction of ammonia more completely in the catalytic decomposition of AP. Thermal gravity analysis-Fourier transform infrared (TGA-FTIR), as a special technic, was used to analyze catalytic mechanism of the thermal decomposition of AP by real-time detecting the products during the decomposition process.
Experimental section
Chemicals and apparatus
Dicyandiamide (99%), perchloric acid (99.999%) and AP (AR) were obtained from Aladdin (Shanghai, China). Program temperature furnace was obtained from Samsung instrument (Xiangtan, China). The mortar was obtained from Xn Nets (Shanghai, China). All reagents are of analytical grade and used without further purification.Preparation of g-C3N4
g-C3N4 was synthesized by previously reported method.10 Briefly, 3 g dicyandiamide was added into the furnace with three heating step. In the first step, the furnace temperature was operated at a heating rate of 10 °C min−1 in air over a temperature range of 25–300 °C. And then at a heating rate of 5 °C min−1 over a temperature range of 300–550 °C. Last step, furnace temperature was kept at 550 °C for 4 h. After naturally cooled to room temperature, g-C3N4 with a faint yellow colour was obtained. Afterwards, the g-C3N4 was grinding about 10 min to obtained ultra-fined powered in the mortar.Sample characterization
Powder X-ray diffraction (XRD) patterns were collected from the prepared samples on a Philips X'Pert Pro X-ray diffractometer (PANalytical, Holland) employing Cu Kα1 radiation (λ = 0.15418 nm). The detailed testing conditions were described as follows: the scan rate (2θ) was 0.05° s−1, the accelerating voltage was 40 kV, and the applied current was 80 mA. Field-emission scanning electron microscopy (FESEM) measurements were performed on an Ultra 55 microscope (ZEISS Company, The German) with an acceleration voltage of 15.0 kV. Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet-5700 FTIR spectrometer using pressed KBr pellets to test the chemical bonding of the samples from 4000 to 225 cm−1. UV-vis spectra were measured on a UV-3150 of ultra-violet visible-near infrared spectrophotometer (Shimadzu, Japan). X-ray photoelectron spectroscopy (XPS) was performed with an ESCALAB 250 electron spectrometer using Al Kα irradiation.Catalytic measurements
To test the catalytic effect of g-C3N4 materials on the decomposition of AP, AP (AR, d50: 135 μm) and g-C3N4 materials with different weight ratios were mixed for 30 min. Then the resulting mixture was detected by thermo-gravimetric analysis/differential scanning calorimetry (TGA-DSC) using Mettler Toledo TGA-DSC1-1100LF at a heating rate of 10 °C min−1 in a static N2 atmosphere over the temperature range of 25–500 °C with Al2O3 as reference. The contents of g-C3N4 hybrids used in AP were 1, 3, 5 and 10 wt%, respectively.Results and discussion
Preparation and characterization of g-C3N4
The g-C3N4 was synthesized by calcining the dicyandiamide via the previously reported approach.10 According to the weight (2.12 g) of product, the calculated yield was 70.6%. The morphology of the prepared g-C3N4 was observed by FESEM. As shown in Fig. 1a, The FESEM image of g-C3N4 shows obviously layered structure, indicating the g-C3N4 consisted of graphitic planes stacking along the c-axis.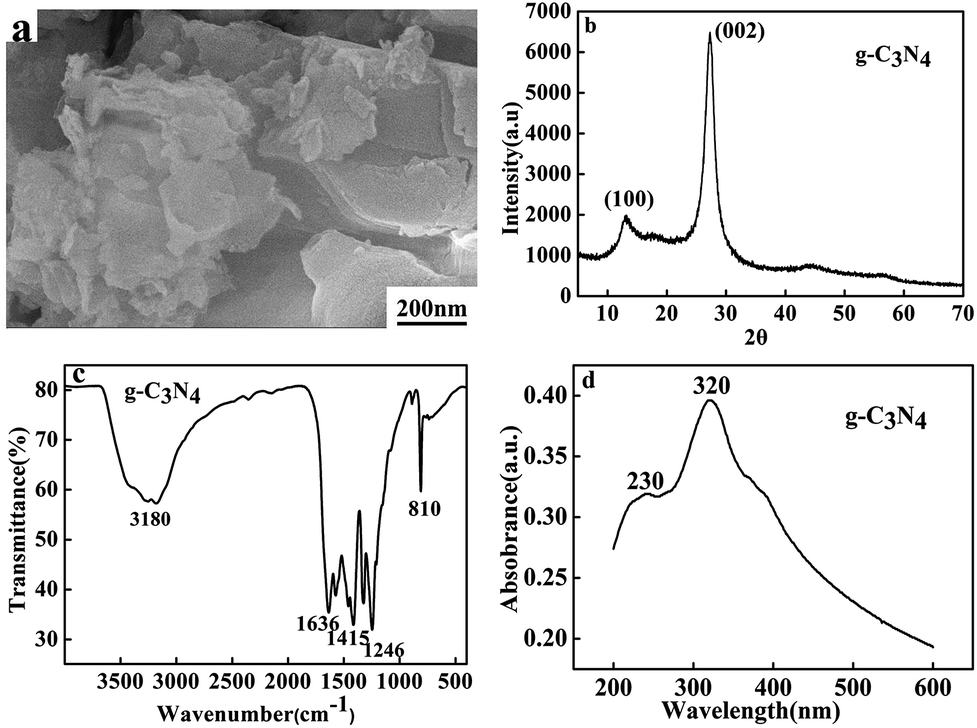 | ||
| Fig. 1 FESEM image (a), XRD pattern (b), FT-IR spectrum (c), and UV-vis spectrum of g-C3N4 (d). | ||
The XRD pattern of g-C3N4 is presented in Fig. 1b. It can be clearly seen that typical XRD peaks of bulk g-C3N4 can be observed and all diffraction peaks are in good agreement with the hexagonal nitride carbon structure with the lattice constants a = 0.4742 nm and c = 6.7205 nm, which is in accord with the literature values of JCPDS no. 87-1526. The strong XRD peak at 27.50° is originated from the (002) interlayer diffraction of graphitic-like structures, corresponding to an interlayer distance of d = 0.326 nm.12 The low angle diffraction peak at 13.3° (d = 0.663 nm), which derived from in-planar repeated triazine units.
Fig. 1c shows the typical FT-IR spectrum of the as-prepared g-C3N4. The sharp band at 810 cm−1 is ascribed to the breathing of the triazine units. The peaks at 1246, 1415, and 1636 cm−1 correspond to the typical stretching modes of CN heterocycles, demonstrating the formation of extended networks of C–N–C bonds.14 The broad bands between 3000 and 3400 cm−1 are attributed to the secondary and primary amines.23 The UV-vis spectrum of g-C3N4 is shown in Fig. 1d. The adsorption of amino peak is located at 230 nm and the strong absorption band at 320 nm was typical of carbon and nitride bond in conjugation.16,17 All the results demonstrated that g-C3N4 has been successfully prepared.
Thermal decomposition of AP catalyzed by g-C3N4
Different amounts of g-C3N4 are mixed as additives with AP to study the catalytic behaviour on the thermal decomposition of AP. The catalytic performance of g-C3N4 in the thermal decomposition of AP was investigated by TGA-DSC measurements as shown in Fig. 2.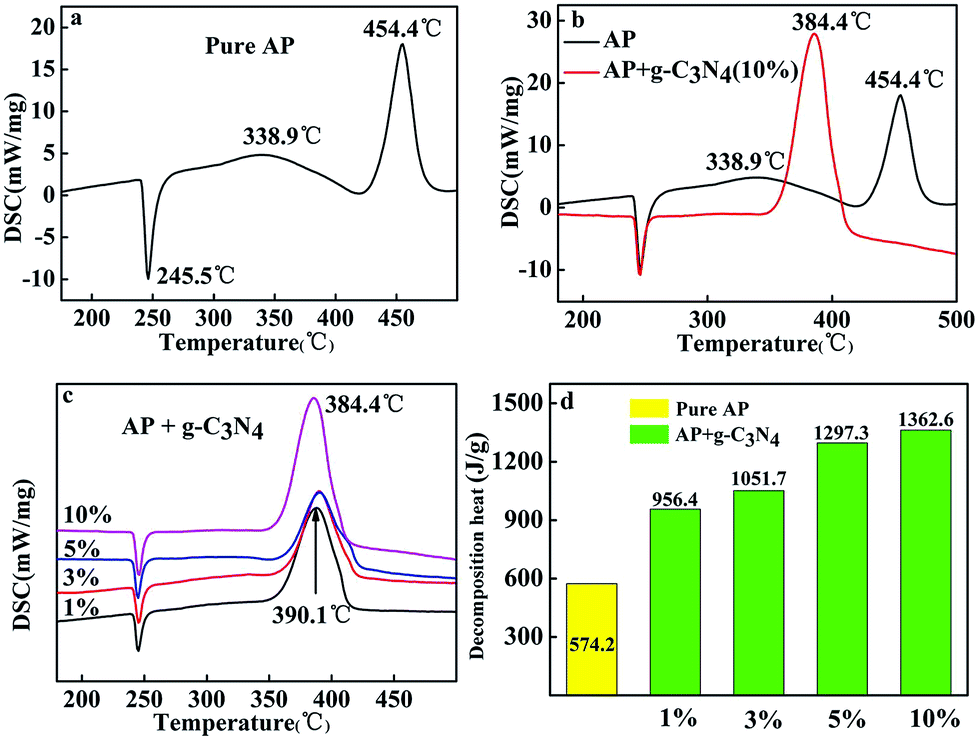 | ||
| Fig. 2 DSC curves of pure AP (a), AP mixed with 10 wt% as prepared g-C3N4 at a heating rate of 10 °C min−1 (b), AP mixed with g-C3N4 (1 wt%, 3 wt%, 5 wt%, 10 wt%) at a heating rate of 10 °C min−1 (c), the catalytic effect comparison of no catalyst and g-C3N4 (d). | ||
Fig. 2a shows the DSC curve of the decomposition of pure AP. Three peaks centred at 245.5, 338.9 and 454.4 °C can be observed. The endothermic peak at 245.5 °C is attributed to the crystallographic transition of AP from orthorhombic to cubic.1 Two exothermic peaks at 338.9 and 454.4 °C are ascribed to the low-temperature thermal decomposition (LTD) and the HTD, respectively.2,24,25 However, when g-C3N4 was added to AP, the cases were apparently changed. As shown in Fig. 2c, with the addition of the g-C3N4, HTD process of AP disappeared to show a sole exothermic process in the temperature range from 384.4–390.1 °C, indicating that the AP thermal decomposition rate was enhanced, while no change was observed for the phase transition temperature of AP. Compared with the HTD temperature of pure AP, the decomposition temperature of the sample with addition of 10 wt% decrease by 70 °C. From the Fig. 2b (DSC curves), two exothermic peaks are clearly observed for pure AP from 250 to 500 °C, while only one exothermic peak is presented for the mixture of AP with 10 wt% g-C3N4, which located at 384.4 °C.
Moreover, with increasing the amounts of g-C3N4 in AP, the decomposition heat reduced, accompanying by a decrease in the maximum decomposition temperature (Fig. 2c). The decomposition heat of AP in the absence and presence of g-C3N4 (10 wt%) were determined according to the integral area of the exothermic process.26 The exothermic heat of AP in the presence of 10 wt% g-C3N4 is about 1362.6 J g−1, which is much higher than that of pure AP (574.2 J g−1, Fig. 2d).
In order to further study catalytic properties of the g-C3N4 for AP thermal decomposition, g-C3N4 was premixed with AP in a mass ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 for DSC tests at different heating rates (Fig. 3). From the exothermic peak temperature as a function of heating rate, several important kinetic parameters for AP decomposition with g-C3N4 additives can be calculated. The relationship between decomposition temperature of AP and heating rate can be described by the Kissinger correlation5,27 in the eqn (1).
:9 for DSC tests at different heating rates (Fig. 3). From the exothermic peak temperature as a function of heating rate, several important kinetic parameters for AP decomposition with g-C3N4 additives can be calculated. The relationship between decomposition temperature of AP and heating rate can be described by the Kissinger correlation5,27 in the eqn (1).
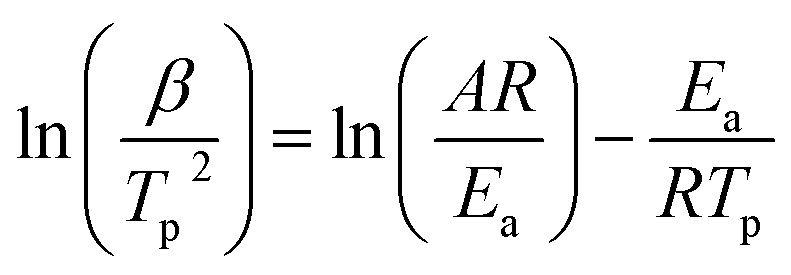 | (1) |
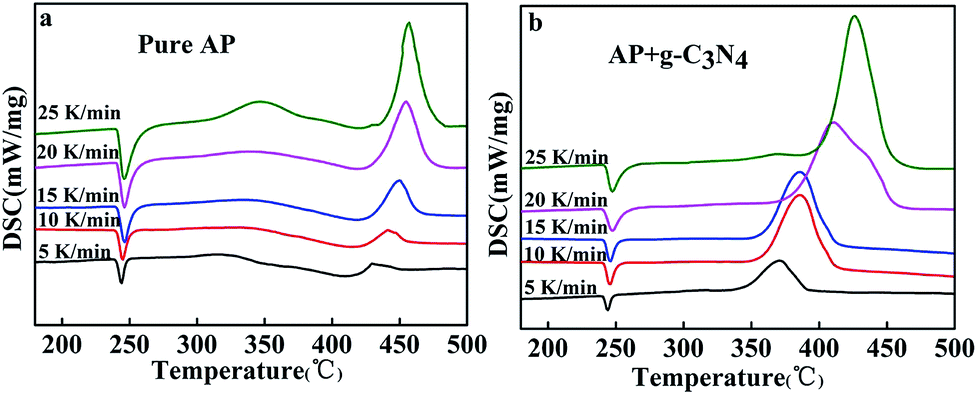 | ||
| Fig. 3 DSC curves of pure AP (a) and AP mixed with 10 wt% as-prepared g-C3N4 (b) at different heating rates. | ||
Fig. 4 shows the experimentally measured ln(β/Tp2) versus 1/Tp with and without g-C3N4 additives. For pure AP, the activation energy of HTD was calculated to be 216.0 kJ mol−1, which is close to the value previously reported in the literature.6 However, the activation energy of AP decomposition in the presence of g-C3N4 additives becomes as small as 119.8 kJ mol−1. Therefore, g-C3N4 as a unique metal free substance in promoting ammonium perchlorate decomposition was one of excellent catalysts (Fig. S3, ESI†).
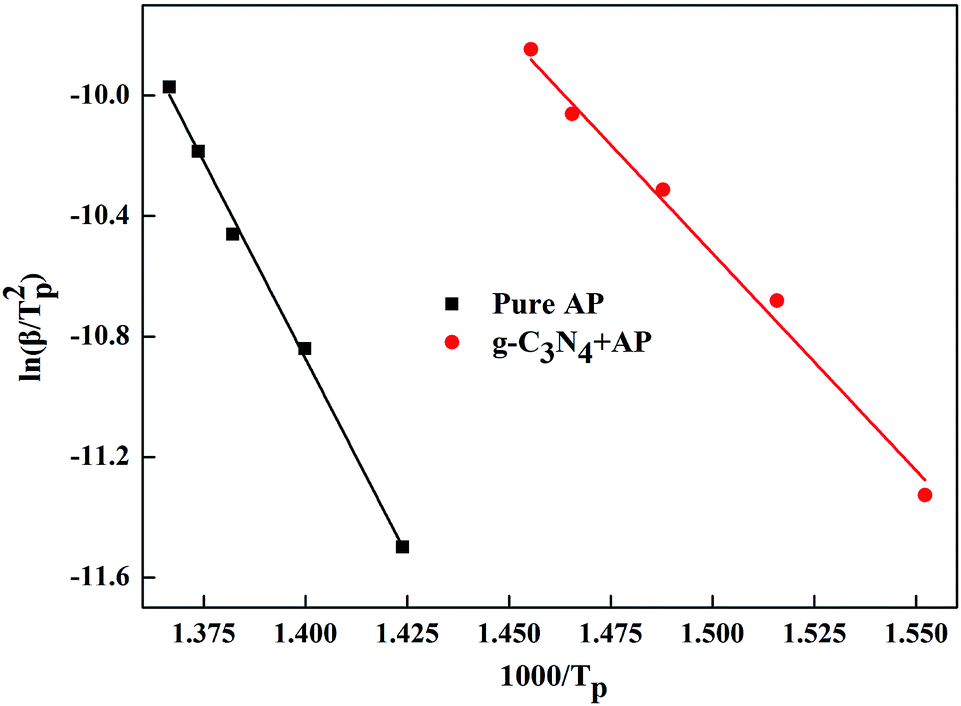 | ||
| Fig. 4 Dependence of ln(β/Tp2) on 1/Tp for AP and mixtures of AP with 10 wt% g-C3N4 additives. Scatter points are experimental data and lines denotes the linear fitting results. | ||
Catalytic mechanism of g-C3N4 for AP thermal decomposition
In order to further identify the role of g-C3N4 in AP thermal decomposition, the morphology and surface structure of g-C3N4 before and after reaction were studied by FESEM, FT-IR, UV-vis and XPS spectra. Fig. 5 shows the FESEM images of the g-C3N4 sample (a) and g-C3N4 residual (b). It can be seen that the morphology of g-C3N4 was not obviously changed after reaction. Furthermore, the purities of g-C3N4 sample was confirmed by the XPS date (Fig. S1†). In addition, according to the fact that the FT-IR spectra, UV-vis absorption, and XPS spectra28 (Fig. 6) of g-C3N4 was almost the same before and after reaction, confirming that g-C3N4 acted as a catalyst and is not damaged after reaction with at least 10 times the amount of ammonium perchlorate. | ||
| Fig. 5 FESEM images of g-C3N4 sample (a) and g-C3N4 residual (b). | ||
 | ||
| Fig. 6 FT-IR spectra (a), UV-vis spectra (b) of g-C3N4 and g-C3N4 residual. | ||
On the basis of the related references, a possible mechanism of thermal decomposition of AP was proposed.
The mechanism of AP thermal decomposition was firstly proposed by Jacobs.29 The first decomposition step of AP involved a solid–gas multiphase reaction containing LTD and HTD process as follows:26
 | (2) |
| NH3(a) + HClO4(a) ⇌ NH3(g) + HClO4(g) (HTD) | (3) |
NH4+ClO4− corresponding to the pair of ions (NH4+ and ClO4−) in the AP lattice. The LTD has been demonstrated to be a heterogeneous process, including a proton transfer from NH4+ to ClO4− to form NH3 and HClO4, the adsorption of NH3 and HClO4 in the porous structure,5 and finally the decomposition of HClO4 and reaction with NH3.29,30 Alternatively, the high-temperature exothermic process is ascribed to the oxidation of NH3 by HClO4 in the gas phase.1,26,31
Why could g-C3N4 catalyze the AP thermal decomposition? It was reported that the activation energy of AP decomposition was related to the LID and HTD step, which changed appreciably in the catalysed systems, indicating that g-C3N4 influenced the primary dissociation of AP into NH3 and HClO4.
Meanwhile, HClO4 was the key chain carriers in the decomposition of AP in its primary stages because it was adsorbed in the pores of AP and prevented the continuous decomposition of AP.32 The unique surface structure of g-C3N4 was consisted of triazine units connected by planar amino groups (Fig. 7), which can be considered as a Lewis base. Thus, HClO4 will be absorbed on the surface of g-C3N4 via the Lewis acid–base interaction (Fig. S4, ESI†), leading to the shift of the equilibrium to the right-hand side (eqn (2)). Thus the Lewis acid–base interaction between g-C3N4 and HClO4 might decrease the activation energy, leading to the acceleration of thermal decomposition of AP.
 | ||
| Fig. 7 Triazine-based connection pattern of g-C3N4. | ||
On the other hand, g-C3N4 has a band gap of approximately 2.7 eV (ref. 17) (Fig. S2†) with a conduction band potential at −1.3 eV vs. RHE, which is easy to meet the requirements of thermal excitation.16 Therefore, the conduction-band electrons (ecb−) and valence band holes (h+) could be generated on the surface of g-C3N4 when g-C3N4 was excited by a heating energy greater than the band gap energy.13,14 During the catalytic process, the generated electrons could react with HClO4 molecular, reducing it to a superoxide radical anion ˙O2−. The thermo-generated ˙O2− and h+ have powerful oxidation ability, which could further react with NH3 to form H2O, NO2 and N2O.17
For the purpose to detect the products of thermal decomposition of AP, TGA-FTIR, as a special analytical instrument, was used to analyze the whole decomposition process by real-time monitoring of FTIR. Three-dimensional FTIR spectra was shown in Fig. 8. Fig. 9 shows the TGA curve of AP decomposition with the 4 wt% additives of g-C3N4.
 | ||
| Fig. 8 Three-dimensional TGA-FTIR spectra of the decomposition products of AP during thermal decomposition. | ||
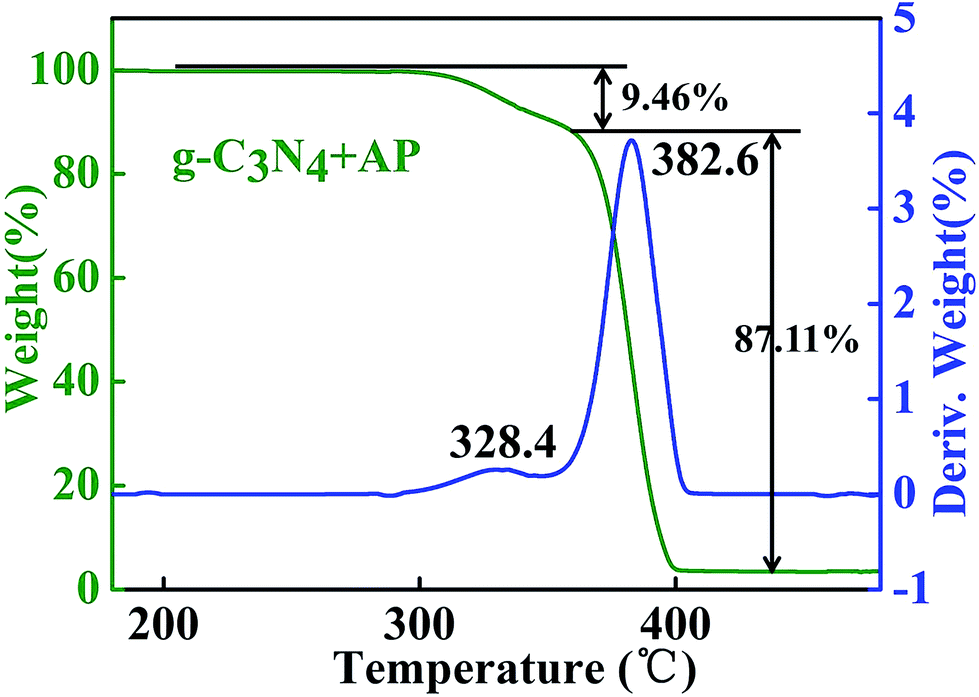 | ||
| Fig. 9 TGA curve of the AP with 4 wt% additives of g-C3N4. | ||
The peaks shifted to a higher temperature than that of the corresponding DTG curve because of the delay time between the gas generation and its detection by the FTIR instrument.
In the TGA curve of AP, two characteristic weight loss step are correspond to the LTD and HTD stages which weight loss rate reach to 9.49 and 87.11%, respectively. The peak temperature of AP decomposition, 382.6 °C, has a good relationship with three-dimensional FTIR spectrum with the strongest peak position.
Fig. 10 shows the FTIR spectra of the gas products during decomposition at their respective peaks. All of the evolved products result from the two decomposition stages of LTD and HTD segment (310–350 and 350–400 °C). The gas product at 408 °C were identified as H2O, NH3 (3400–3650 and 1650–1620 cm−1), HCl (1750–3000 cm−1), N2O, NO2 (2202–2238, 1380–1320 and 840–800 cm−1) and ClO3 (1000–900 cm−1). In addition, the gas products of decomposition process at 418, 378, 358 and 298 °C were also confirmed the existence of the above gas. The date of TGA-FTIR spectroscopy showed that the main gas products of AP were H2O, HCl, N2O, NO2 and ClO3.
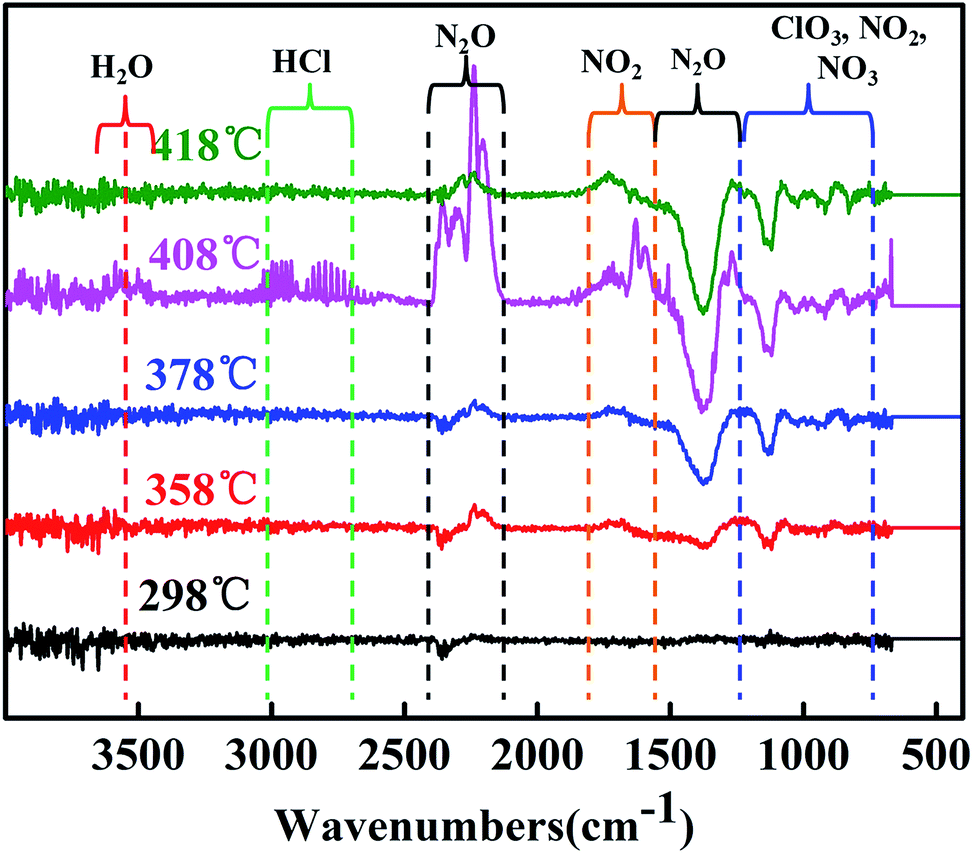 | ||
| Fig. 10 FTIR spectra of gas products during decomposition at respective peaks. | ||
Meanwhile, a series of reactions increasing the exothermic heat of the thermal decomposition process occurred on the surface of g-C3N4. Thus, g-C3N4 not only decreased the activation energy of AP decomposition during the LID and HTD step,2,5 but also promoted the reduction of HClO4 and the oxidation of NH3, so that the decomposition temperature of AP reduced, or even the decomposition stage concentrated in one step, and the heat release also increased.
In brief, the thermal decomposition reaction of AP in the presence of g-C3N4 as the catalyst may proceed as shown in Fig. 11.
 | ||
| Fig. 11 Schematic of the thermal decomposition process of AP with g-C3N4. | ||
Conclusions
In summary, g-C3N4 has been proved to be a metal-free and environmentally friendly catalyst for thermal decomposition of AP for the first time. The HTD temperature of AP apparently decreased followed by a significant increase in the decomposition heat. Notably, the catalytic performance was directly related to the g-C3N4 content. A possible catalytic mechanism of g-C3N4 for AP thermal decomposition was also proposed. The triazine units connected by planar amino groups of g-C3N4 could be favorable for the adsorption and diffusion of perchloric acid via Lewis acid–base interactions, which decreased the activation energy of AP decomposition. And the semiconducting g-C3N4 produced ecb− and h+ that promoted the reduction of HClO4 and the oxidation of NH3, which boosted the AP thermal decomposition. The developed catalyst for AP thermal decomposition had several distinctive advantages: (1) the preparation process of g-C3N4 was simple, easy to perform, and cost-effective; (2) g-C3N4 did not contain any metal elements or poisonous substance, thus it was metal-free and environmentally friendly. These advantages made g-C3N4 promising as a low cost, metal-free, and environmentally friendly modifier in AP-based composite solid rocket propellants.Acknowledgements
This work was supported by the Natural Science Foundation of China (51372211, 10576206 and 21301142), and Defence Science and Technology Project (A3120133002) and Carbon nanomaterial's research team in Sichuan Youth Science and Technology Innovation Special (2011JTD0017) for the State Key Laboratory Cultivation Base for Non-metal Composites and Functional Materials, Southwest University of Science and Technology. The authors thank China Academy of Engineering Physics for their assistance in the XPS characterization. Also, the technology was supported by the Analytical and Testing Center of SWUST for performing XRD and FESEM characterizations.Notes and references
- V. Boldyrev, Thermochim. Acta, 2006, 443, 1 CrossRef CAS PubMed
.
- S. Vyazovkin and C. A. Wight, Chem. Mater., 1999, 11, 3386 CrossRef CAS
- N. Li, Z. Geng, M. Cao, L. Ren, X. Zhao and B. Liu, Carbon, 2013, 54, 124 CrossRef CAS PubMed
- D. Zhang, Q. Xie, A. Chen, M. Wang, S. Li and X. Zhang, Solid State Ionics, 2010, 181, 1462 CrossRef CAS PubMed
- L. Li, X. Sun, X. Qiu, J. Xu and G. Li, Inorg. Chem., 2008, 47, 8839 CrossRef CAS PubMed
- M. Zou, X. Jiang, L. Lu and X. Wang, J. Hazard. Mater., 2012, 225–226, 124 CrossRef CAS PubMed
- X. Guan, L. Li, J. Zheng and G. Li, RSC Adv., 2011, 1, 1808 RSC
- R. A. Chandru, S. Patra, C. Oommen, N. Munichandraiah and B. N. Raghunandan, J. Mater. Chem., 2012, 22, 6536 RSC
- W. Zhang, P. Li, H. Xu, R. Sun, P. Qing and Y. Zhang, J. Hazard. Mater., 2014, 268, 273 CrossRef CAS PubMed
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen and J. D. Epping, Angew. Chem., Int. Ed., 2010, 49, 441 CrossRef CAS PubMed
- M. Kazuhiko, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940 Search PubMed
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin and J. M. Carlsson, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed
- L. Zhang, D. Jing, X. She, H. Liu, D. Yang and Y. Lu, J. Mater. Chem. A, 2014, 2, 2071–2078 CAS
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. mater., 2009, 21, 1609 CrossRef CAS
- J. Zhang, B. Wang and X. Wang, Progr. Chem., 2014, 26, 19 CAS
- A. M. Stoneham, Rep. Prog. Phys., 1981, 44, 1251 CrossRef
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596 CrossRef CAS
- J. Kouvetakis, M. Todd and B. Wilkens, Chem. Mater., 1994, 6, 811 CrossRef CAS
- T. Sekine, H. Kanda, Y. Bando, M. Yokoyama and K. Hojou, J. Mater. Sci. Lett., 1990, 9, 1376 CrossRef CAS
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Müller and R. Schlögl, J. Mater. Chem., 2008, 18, 4893 RSC
- M. R. Wixom, J. Am. Ceram. Soc., 1990, 73, 1973 CrossRef CAS PubMed
- M. J. Bojdys, J. O. Muller, M. Antonietti and A. Thomas, Chem.–Eur. J., 2008, 14, 8177 CrossRef CAS PubMed
- T. Ma, Y. Tang, S. Dai and Q. Qiao, Small, 2014, 10, 2382 CrossRef CAS PubMed
- D. L. Reid, A. E. Russo, R. V. Carro, M. A. Stephens, A. R. LePage and T. C. Spalding, Nano Lett., 2007, 7, 2157 CrossRef CAS
- Z. Liu, C. Yin, Y. Kong, F. Zhao, Y. Luo and H. Xiang, Energ. Mater., 2000, 8, 75 CAS
- G. Tang, S. Tian, Z. Zhou, Y. Wen, A. Pang and Y. Zhang, J. Phys. Chem. C, 2014, 118, 11833 CAS
- Q. Yang, S. Chen, G. Xie and S. Gao, J. Hazard. Mater., 2011, 197, 199 CrossRef CAS PubMed
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen and Z. Guo, Angew. Chem., Int. Ed., 2014, 53, 9240 CrossRef CAS PubMed
- P. W. M. Jacobs and A. R. Jones, J. Phys. Chem., 1968, 72, 202 CrossRef CAS
- X. Sun, X. Qing, L. Li and G. Li, Inorg. Chem., 2008, 47, 4146 CrossRef CAS PubMed
- E. F. Khairetdinov and V. V. Boldyrev, Thermochim. Acta, 1980, 41, 63 CrossRef CAS
- Z. Guo, D. Chen and C. J. Lee, J. Phys. Chem. B, 2006, 110, 15689 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01157d |
| This journal is © The Royal Society of Chemistry 2015 |