
L-Amino acid derived pyridinium-based chiral compounds and their efficacy in chiral recognition of lactate†
Kumaresh Ghosh* and
Anupam Majumdar
Department of Chemistry, University of Kalyani, Kalyani-741235, India. E-mail: ghosh_k2003@yahoo.co.in; Fax: +91 3325828282; Tel: +91 3325828750 ext. 306
First published on 20th February 2015
Abstract
A series of pyridinium-based chiral compounds 1–6 have been designed and synthesized. L-Amino acids have been used as the chiral source in the molecules. Among the chiral compounds, an L-valine derived compound 1 was found to exhibit enantioselective recognition of D-lactate in fluorescence. Structural tuning of this derivative, either by replacing L-valine with L-alanine or L-phenylglycine or by reducing the number of chiral centres around the binding site, does not result in any significant change in enantioselectivity in the recognition process. Change of the urea site to amide introduces compound 6 that displays good enantiodiscrimination for lactate (enantiomeric fluorescence difference ratio ef = 28.33 for D-lactate), even better than that of the L-valine derived compound 1 and of other reported structures in the literature.
Introduction
Enantioselective recognition of chiral analytes is an important topic in chiral recognition.1 Chiral recognition is unavoidably significant as chiral biomolecules such as proteins, nucleic acids and carbohydrates play a vital role in life. In this area, synthetic receptors that are capable of discriminating a particular isomer from its mirror image isomer through photophysical changes have attracted much attention. Of the different photophysical processes, fluorescence is noteworthy because of high sensitivity and thus its application in chiral recognition has been studied for several decades.2 Chiral fluorescence-based sensors can be used in the rapid determination of enantiomeric composition as well as in high-throughput catalysis for asymmetric synthesis.3 Thus the development of fluorescence-based enantioselective sensors for distinguishing chiral amines,4a amino alcohols,4b–c chiral acids,4d chiral amino acids4e–h and carbohydrates4i–j etc., has begun to attract research attention. In past several years, enantioselective recognition of biologically significant α-hydroxycarboxylic acids5 as well as of α-amino acids and their derivatives4e–h has been much explored. Of the different α-hydroxycarboxylates, lactate is a simple example which is biologically relevant. While the isomer L(+)-lactate is important in biological metabolism, D(−)-lactate is harmful to human metabolism and can result in acidosis and decalcification.6 Thus the enantioselective binding of lactate or its derivatives is crucial, although there are few reports in the literature.5g,7 Recently, we have reported L-valine derived pyridinium-based chiral receptor 1, which shows enantioselective binding of lactate by exhibiting significant change in emission in the presence of D-lactate over L-lactate in CH3CN.8 In relation to this, we wish to report in this full account a series of pyridinium-based chiral hydrogen bonding receptors 2–6 and their chiral recognition properties towards hydroxycarboxylates in detail, emphasizing the importance of structural tuning to achieve good chiral discrimination.
It is to be pointed out that the exploration of 3-aminopyridine in devising chiral sensors for the enantioselective recognition of carboxylate-based substrates is unknown in the literature except for our recent example of structure 1.8
Results and discussion
Compounds 1–6 were synthesized according to Schemes 1 and 2. Following our reported reaction protocols for the synthesis of 1,8 compounds 2 and 3 were synthesized (Scheme 1). Boc protected L-amino acids such as valine 7a, alanine 7b and phenyl glycine 7c were reacted with 3-aminopyridine in the presence of dicyclohexylcarbodiimide (DCC) to give the coupled products 8a–c, respectively. Removal of the Boc-groups in 8a, 8b and 8c using trifluoroacetic acid (TFA) gave the amines 9a, 9b and 9c, respectively, which individually upon reaction with 1-naphthyl isocyanate, obtained from the reaction of 1-naphthylamine with triphosgene, yielded the respective urea derivatives 10a, 10b and 10c. For quarternization of the pyridine ring nitrogens in 10a–c, the chloroamides 11a–c, which were obtained from the reactions of methyl esters of amino acids with chloroacetyl chloride, were used. Individual reflux of compounds 10a–c in the presence of the corresponding chloroamides 11a–c in dry CH3CN for 4 days afforded the chloride salts of 1–3. Next, anion exchange reactions of the chloride salts of 1–3 were pursued using NH4PF6 to give the desired compounds 1, 2 and 3. On the other hand, reaction of 10a with benzyl bromide in dry CH3CN followed by Br− exchange with PF6− gave compound 4. | ||
| Scheme 1 (i) 3-Aminopyridine, DCC, CH2Cl2, 20 h; (ii) TFA, CH2Cl2, 3 h; (iii) 1-naphthylamine, triphosgene, Et3N, CH2Cl2, 16 h; (iv) (a) 11a–c, CH3CN, reflux, 4 days; (b) NH4PF6, CH3OH–H2O; (v) (a) benzyl bromide, CH3CN, reflux, 18 h; (b) NH4PF6, CH3OH–H2O. | ||
 | ||
| Scheme 2 (a) (i) 1-Naphthylamine, triphosgene, Et3N, CH2Cl2, 18 h; (ii) 11a, CH3CN, reflux, 4 days; (iii) NH4PF6, CH3OH–H2O; (b) (iv) 1-naphthylacetyl chloride, CH2Cl2, Et3N, 12 h; (v) 11a, CH3CN, reflux, 3 days; (vi) NH4PF6, CH3OH–H2O. | ||
Compounds 5 and 6 were synthesized according to Scheme 2. Triphosgene mediated reaction of 3-aminopyridine, followed by addition of 1-naphthylamine in Scheme 2a gave the urea 12, which on further reaction with the chloroamide 11a, gave the chloride salt 13. Chloride exchange in 13 using NH4PF6 gave the desired compound 5. On the other hand, the amine 9a was coupled with 1-naphthylacetyl chloride to give the amide derivative 14, which, under reflux in CH3CN in the presence of chloroamide 11a, gave the chloride salt 15. Next, chloride exchange in 15 using NH4PF6 gave the desired compound 6 in appreciable yield. All the compounds were characterized by usual spectroscopic methods.
Chiral recognition requires multiple-point interaction.9 Analysis of the structures 1–6 reveals that the pyridinium cation is the principal binding site in all cases. Around this motif, other different functional groups have been considered in different ways to have diverse chiral receptor structures that are capable of attaining multiple-point interaction. Variation of the amino acid in the design strategy has been considered to account for the steric requirements in the binding site for good chiral discrimination. The naphthalene moiety has been used as the fluorescence signaling unit to assess the chiral recognition behavior of the molecules. It is to be pointed out that the 3-aminopyridinium motif is important in anion recognition as it provides hydrogen bond donors from different anchoring groups and also a polarized C–H bond at the ortho position to the anion and the complex is further stabilized by charge–charge interaction. Its widespread use in anion recognition by different researchers and also by us has recently been thoroughly reviewed.10
To study the chiral recognition properties of the receptor structures 1–6, tetrabutylammonium salts of D- and L-hydroxy acids such as lactic and mandelic acids were used. In our earlier report8 we showed that compound 1 selectively recognizes D-lactate over L-lactate with an ef [ef = (ID − I0)/(IL − I0) where I0 represents the fluorescence emission intensity in the absence of the chiral substrate, IL and ID are the fluorescence intensities in the presence of L- and D-lactates, respectively] of 5.32. To understand the structural role of L-valine (chiral source) in 1, we studied compound 2, which bears L-alanine instead of L-valine. The fluorescence study of 2 (c = 3 × 10−5 M) in CH3CN revealed poor enantiodiscrimination. Upon gradual addition of guests to the solution of 2 in CH3CN, the emission of 2 at 378 nm increased with a red shift of ∼28 nm. But the change in emission for each pair occurred to nearly the same extent (Fig. S1, ESI†). Only in the case of D-lactate it was slightly more (ef = 1.16). Fig. 1, in this regard, shows the change in emission of 2 in the presence of 20 equiv. of D/L-lactates and mandelates in CH3CN. As can be seen from Fig. 1, receptor structure 2 shows preferential binding to the isomers of lactates than to mandelate. But the enantioselection was insignificant. This was also true in the ground state. In a UV-vis study, no marked difference in absorption spectra of 2 during titration was observed (Fig. S2, ESI†).
 | ||
| Fig. 1 Change in fluorescence ratio of 2 (c = 3 × 10−5 M) upon addition of 20 equiv. of guests in CH3CN at 406 nm (λexc = 300 nm). | ||
Under identical conditions, a fluorescence study of 3 (c = 3 × 10−5 M), which contains L-phenyl glycine as the chiral source, also revealed poor enantiodiscrimination in CH3CN. On addition of the guests to a solution of 3 in CH3CN the emission of 3 at ∼400 nm increased (Fig. S3, ESI†) to the same extent and in the case of L-lactate it was a little more (ef = 1.08). Fig. 2 represents the change in emission ratio of 3 in the presence of 6 equiv. of D/L-lactates and mandelates in CH3CN. Further addition of guests to the receptor solution decreased the emission gradually (Fig. S3, ESI†). Similar to 2, in UV titration no characteristic difference in the absorption spectra of 3 was observed in the presence of the guests (Fig. S4, ESI†).
 | ||
| Fig. 2 Change in fluorescence ratio of 3 (c = 3 × 10−5 M) upon addition of 6 equiv. of guests in CH3CN at 405 nm (λexc = 300 nm). | ||
Thus, these results corroborate that in the present study, valine with its steric isopropyl substituent at the α-carbon is superior to alanine and phenyl glycine with methyl and phenyl groups, respectively, in chiral recognition to discriminate the enantiomers of lactate.
After establishing L-valine as the suitable chiral source in the series, we moved further to understand its positional role around the pyridinium motif in the chiral discrimination of α-hydroxycarboxylate. For this, compounds 4 and 5 were synthesized. While in 4 the L-valine unit is present at the 3-position of the pyridinium motif, in compound 5 it is present on the pyridinium ring nitrogen. Fluorescence titrations of these two compounds in CH3CN revealed that both 4 and 5 were unable to show chiral discrimination of the guests studied. In the case of receptor 4, although the lactate-induced change in emission was greater than the cases with mandelates, the enantiomers of lactate were discriminated with ef = 1.65 (Fig. 3a). In comparison, chiral receptor 5 under identical conditions in CH3CN did not exhibit any fluorescence selectivity between lactate and mandelate. But the enantiomers of lactate were poorly discriminated with an ef of 1.18 (Fig. 3b). These observations underline the fact that the presence of L-valine as a single unit either at the ring nitrogen or at the amine function of 3-aminopyridine does not do much to cause enantiodiscrimination of lactate.
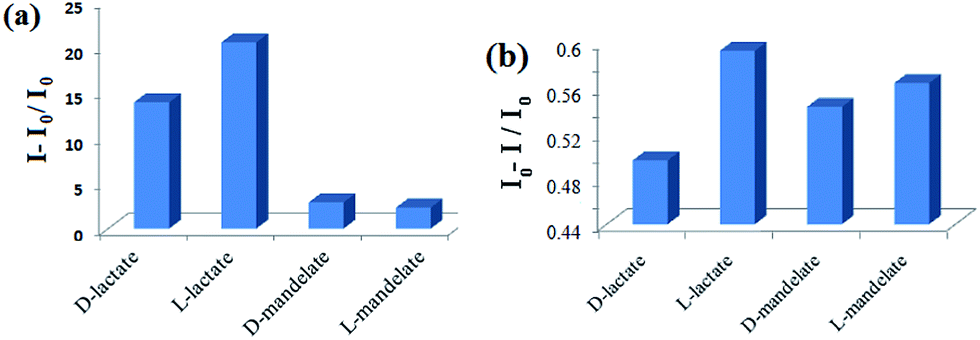 | ||
| Fig. 3 Change in fluorescence ratio of (a) 4 (c = 3 × 10−5 M) upon addition of 13 equiv. of guests in CH3CN at 402 nm (λexc = 300 nm), and (b) 5 (c = 3 × 10−5 M) upon addition of 19 equiv. of guests in CH3CN at 401 nm (λexc = 300 nm). | ||
To evaluate the potentiality of the urea functionality in the design, we then considered amide analogue 6. A thorough fluorescence study of 6 (c = 3 × 10−5 M) was carried out in CH3CN. Like urea analogue 1, compound 6 selectively discriminated the stereoisomers of lactates. Upon gradual addition of D-lactate to a solution of 6 in CH3CN, while the emission at 338 nm decreased to a small extent, a new emission at 425 nm appeared with significant intensity. In comparison, a small increase in emission at 425 nm with the addition of L-lactate was observed. Fig. 4 represents the titration spectra for 6 with both D- and L-lactates. Under identical conditions, titration experiments for 6 with D- and L-mandelates were performed and negligible changes in emission were found. No peak at 425 nm was noticed during interaction (Fig. S9, ESI†). It is thus presumed that the peak at 425 nm in the presence of the lactates is due to the formation of an intermolecular excimer between the naphthalenes.11 Mandelate, being more bulky than lactate, is weakly involved in binding and thus is unable to participate in intermolecular chelation, responsible for excimer emission.
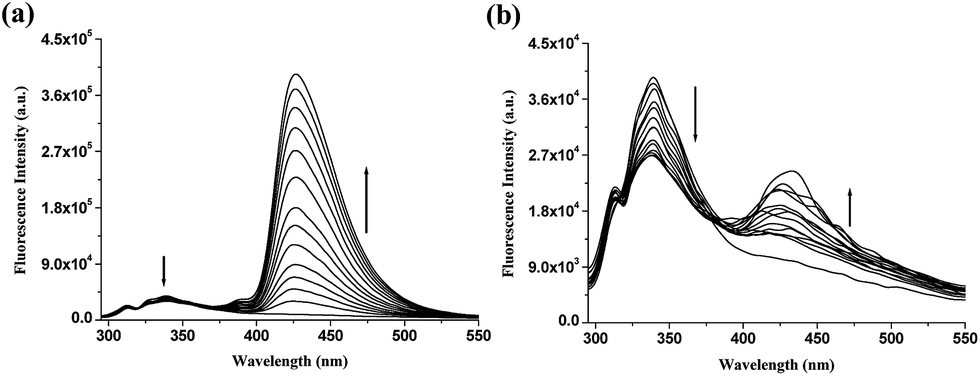 | ||
| Fig. 4 Fluorescence titration spectra for 6 (c = 3 × 10−5 M) in CH3CN with tetrabutylammonium salts of (a) D- and (b) L-lactic acids (in all cases 6 × 10−4 M is a maximal concentration of anion applied; λexc = 285 nm). | ||
Fig. 5 shows the emission ratio of 6 in the presence of D/L-lactates and mandelates in CH3CN. As can be seen from Fig. 5, while the receptor 6 shows sharp fluorometric discrimination between D- and L-lactates at 425 nm, enantiomers of mandelate are scarcely discriminated. The enantiomeric fluorescence difference ratio, ef, is determined to be 28.33, which is considerably greater than the case with 1. Even, the ef value is significantly greater than that for the salen-based chiral fluorescent sensor reported by Song et al.5g It is further to be pointed out that the quantum yield12 of 6 is much more enhanced upon interaction with D-lactate than with L-lactate. The increment is considerable with respect to the receptor 1 (ESI, Table S1†). Thus the amide analogue 6 is established to be more efficient than 1 in the enantioselective recognition of lactate.
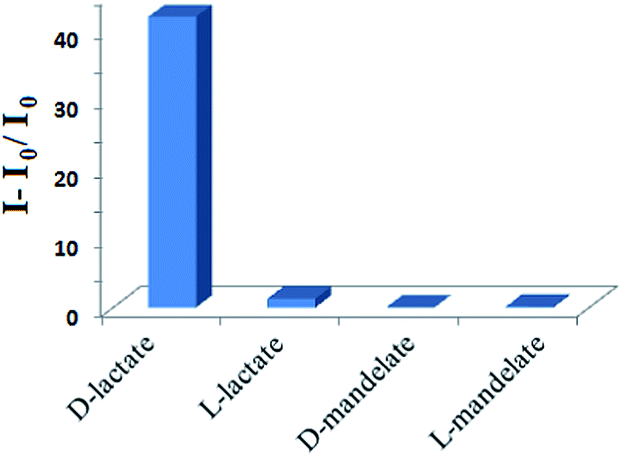 | ||
| Fig. 5 Change in fluorescence ratio of 6 (c = 3 × 10−5 M)) upon addition of 17 equiv. of guests in CH3CN at 425 nm. | ||
In the interaction process, the stoichiometry of the interaction of 6 with both D- and L-lactates was determined to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, as confirmed by Job plot13 (Fig. S11, ESI†). In this context, it is to be pointed out that the appearance of the intermolecular excimer between the naphthalenes in Fig. 4 may originate from the equilibrium species with the stoichiometry of adducts 1:1 (polymeric assembly), 2:2 or others. However, the binding constant value14 was determined from a nonlinear fit of the emission titration data and found to be (1.55 ± 0.19) × 104 M−1 for D-lactate (Fig. 6). For L-lactate, it was determined to be (5.29 ± 0.89) × 103 M−1 (Fig. S12, ESI†) and this is about three times less than the binding constant value for D-lactate. However, due to poor change in emission, we were unable to fit the titration data to a nonlinear equation to determine the binding constant values for both the stereoisomers of mandelates. A comparison of the binding constant values as shown in Table S2 (ESI†) shows that the receptor 6, among the designs, exhibits stronger binding towards D-lactate.
:1, as confirmed by Job plot13 (Fig. S11, ESI†). In this context, it is to be pointed out that the appearance of the intermolecular excimer between the naphthalenes in Fig. 4 may originate from the equilibrium species with the stoichiometry of adducts 1:1 (polymeric assembly), 2:2 or others. However, the binding constant value14 was determined from a nonlinear fit of the emission titration data and found to be (1.55 ± 0.19) × 104 M−1 for D-lactate (Fig. 6). For L-lactate, it was determined to be (5.29 ± 0.89) × 103 M−1 (Fig. S12, ESI†) and this is about three times less than the binding constant value for D-lactate. However, due to poor change in emission, we were unable to fit the titration data to a nonlinear equation to determine the binding constant values for both the stereoisomers of mandelates. A comparison of the binding constant values as shown in Table S2 (ESI†) shows that the receptor 6, among the designs, exhibits stronger binding towards D-lactate.
 | ||
| Fig. 6 Nonlinear curve fitting of the fluorescence titration data for 6 with D-lactate. | ||
However, the enantioselective response of 6 towards a particular stereoisomer of lactate was further realized from the change in emission of 6 in the presence of its mirror image isomer. Fig. 7, in this context, demonstrates these features. L-Lactate induced change in emission of 6 was further perturbed to a considerable extent upon addition of D-lactate (Fig. 7a). The reverse addition was observed to be insignificant (Fig. 7b).
 | ||
| Fig. 7 Fluorescent response of 6 (c = 3 × 10−5 M) to (a) D-lactate (c = 6 × 10−4 M) in the presence of L-lactate (c = 6 × 10−4 M) and (b) L-lactate (c = 6 × 10−4 M) in the presence of D-lactate (c = 6 × 10−4 M) in CH3CN. | ||
For practical applications, we also investigated the chiral recognition behaviour of 6 with the same guests in aqueous CH3CN (CH3CN:H2O = 4:1 v/v). Surprisingly, guest-induced minor change in emission indicated its inefficiency in chiral recognition in aqueous organic solvent (ESI, Fig. S13†).
Prior to studying the interaction of 6 with the lactate isomers via 1H NMR, a NOESY spectrum of 6 was recorded in CD3CN to identify the positions of the different protons (ESI, Fig. S14†). Among the different protons, the interacting protons of 6 were identified in the 1H NMR spectrum by observing the positional movement of the signals in the presence of D- and L-lactates. In relation to this, Fig. 8 highlights the spectral changes of 6 in the presence of D-lactate. All the amide protons of types ‘a’, ‘b’ and ‘c’ underwent downfield chemical shifts in the presence of D-lactate. The amide proton of type ‘a’ became invisible in the presence of a small amount of the guest and this is presumed to be due to its strong participation in hydrogen bonding that makes the signal broaden. On the other hand, while the amide proton of type ‘c’ is significantly shifted downfield, the amide proton of type ‘b’ exhibits a small change in chemical shift upon complexation. The participation of the pyridinium motif in complexation was also recognized from the downfield chemical shifts of the ortho protons of types ‘d’ and ‘e’. The plot in the inset of Fig. 8 shows the change in chemical shifts of the different interacting protons with guest concentration. It is of note that, during interaction, some signals for naphthalene ring protons (asterisks marked in Fig. 8; for details see Fig. S14†) in the regions 7.93 ppm and 7.50 ppm underwent an upfield chemical shift when D-lactate was gradually added. Guest-induced intermolecular chelation of the receptors in solution probably gives a situation where the naphthalene ring experiences π-stacking interaction. This is in accordance with the observation of an excimer emission peak11 at 425 nm in fluorescence (see Fig. 4). A similar observation in the 1H NMR spectra of 6 was noted upon gradual addition of L-lactate, although the changes in chemical shifts of the interacting protons were less compared to those in the case of D-lactate (Fig. S15, ESI†).
 | ||
| Fig. 8 Partial 1H NMR (400 MHz, CD3CN) spectra of 6 (c = 1 × 10−2 M) in the absence and presence of D-lactate [inset: change in chemical shift values for the interacting protons with guest concentration]. | ||
Generally the selectivity of a chiral environment towards the two enantiomers of a chiral species is due to the formation of transient diastereomeric adducts, which differ in both energetic and structural aspects. In the present case, in spite of the several possibilities of stoichiometries of the adducts (1:1, 2:2 or others) in solution, we became interested to study the hydrogen bonding features of the receptor 6 with the lactate isomers and also the corresponding stabilities of the discrete 1:1 adducts in the gas phase through DFT calculations. DFT optimization15 was done in the gas phase using the B3LYP function and 3–21 basis set (Fig. 9). Like structure 1,8 receptor module 6 also showed a preference for D-lactate over its mirror image isomer. D-Lactate is observed to be complexed 4.56 kcal mol−1 more strongly than the L-isomer. Fig. 9 represents the DFT optimized structures in the gas phase with the hydrogen bonding schemes.
 | ||
| Fig. 9 DFT optimized geometries of the complexes of (a) 6 D-lactate (hydrogen bond distances in Å: a = 1.60, b = 1.95, c = 2.19 and d = 1.78); and (b) 6 L-lactate in the gas phase (hydrogen bond distances in Å: a = 1.52, b = 1.92, c = 2.39 and d = 1.79). | ||
Conclusions
In summary, we have designed and synthesized a series of pyridinium-based chiral receptors 1–6 of which structures 1 and 6 have been established as chiral receptors for the enantioselective sensing of D-lactate over its mirror image. Both the structures contain an L-valine unit as the chiral source, which brings good enantioselectivity in the recognition process. Other amino acids such as L-alanine and L-phenyl glycine as the chiral source in the designed structures 4 and 5, respectively, did not bring any enantioselectivity. The steric features of the side chain of the α-amino acid, hydrogen bonding effects and charge–charge interactions play important roles in the recognition process. It is further of note that between 1 and 6, the receptor structure 6, with an amide functionality at the site of the naphthalene instead of a urea, shows greater enantioselectivity (ef = 28.33) towards D-lactate and establishes its efficacy in the chiral recognition of lactates. Further insight along this direction is underway in our laboratory.Experimental
Compounds 8a, 10a, 11a and 1 were obtained according to our earlier reported procedure.8 Synthetic procedures and characterization data of these compounds are further given in the ESI.† Compounds 2, 3 and the intermediates such as 8b and c, 10b and c and 11b and c were prepared according to the same procedure as followed for 1 (ESI†).8Compound (4)
To a stirred solution of 10a (0.1 g, 0.275 mmol) in CH3CN (15 mL), was added benzyl bromide (0.06 g, 0.35 mmol) and the mixture was refluxed for 18 h. After completion of reaction, purification of the crude reaction mixture by preparative TLC using ethyl acetate as eluent gave the bromide salt of 4 as a gummy product (0.11 g, yield: 74%). In the next step, based on the procedure of anion exchange as followed in the case of 1, the bromide ion was exchanged with the PF6− ion using NH4PF6 (0.1 g, 0.61 mmol) in MeOH–H2O (20 mL) to give the desired compound 4 (0.115 g, yield: 93%), mp 120 °C, [α]25D = 19.41 (c = 0.618 g/100 mL, CH3CN), 1H NMR (400 MHz, CDCl3 containing two drops d6-DMSO): δ 10.80 (s, 1H), 9.36 (s, 1H), 8.52 (s, 1H), 8.33 (d, 1H, J = 5.20 Hz), 8.23 (d, 1H, J = 8 Hz), 8.08 (d, 1H, J = 4 Hz), 7.84 (d, 1H, J = 8 Hz), 7.79–7.76 (m, 1H), 7.58–7.52 (m, 2H), 7.46–7.43 (m, 3H), 7.39–7.33 (m, 5H), 7.01 (d, 1H, J = 8 Hz), 5.51 (s, 2H), 4.47 (t, 1H, J = 8 Hz), 2.23–2.18 (m, 1H), 1.08–1.05 (m, 6H); 13C NMR (100 MHz, CDCl3 containing two drops d6-DMSO): δ 173.3, 156.7, 139.8, 138.1, 134.4, 134.06, 134.0, 133.9, 132.0, 130.0, 129.5, 129.0, 128.3, 128.1, 126.8, 125.8, 125.74, 125.70, 123.7, 121.5, 118.7, 65.2, 60.0, 30.9, 19.4, 18.0; FT-IR: ν cm−1 (KBr): 3631, 3367, 3095, 2966, 1707, 1655, 1595, 1547, 1504; HRMS (TOF MS ES+): calcd for (M − PF6)+: 453.2285, found: 453.2233.1-(Naphthalene-1-yl)-3-(pyridine-3-yl)urea (12)
To a stirred solution of triphosgene (0.5 g, 1.68 mmol) in dry CH2Cl2 (5 mL), was added 3-aminopyridine (0.16 g, 1.7 mmol) dissolved in dry CH2Cl2 (25 mL), dropwise using a dropping funnel for 30 min. After complete addition of 3-aminopyridine, triethylamine (0.62 mL, 2.5 equiv.) was added and the reaction mixture was stirred for another 40 min. Then 1-naphthylamine (0.27 g, 1.89 mmol) in dry CH2Cl2 (25 mL), was added to the reaction mixture. Stirring of the reaction mixture was continued for 16 h. After completion of reaction, CH2Cl2 was evaporated off and water was added to the residue. The aqueous layer was extracted with CHCl3 (25 mL × 3) and dried over anhydrous Na2SO4. After evaporation of the solvent, the crude mass was purified by silica gel column chromatography using petroleum ether–ethyl acetate (2:3, v/v) as eluent to give the compound 12 (0.3 g, yield: 68%), mp 221 °C, 1H NMR (400 MHz, d6-DMSO): δ 9.30 (s, 1H), 8.97 (s, 1H), 8.72 (s, 1H), 8.28 (d, 1H, J = 4.80 Hz), 8.19 (d, 1H, J = 8 Hz), 8.09–8.00 (m, 3H), 7.75 (d, 1H, J = 8 Hz), 7.69–7.60 (m, 2H), 7.55 (t, 1H, J = 8 Hz), 7.40 (dd, 1H, J1 = 8 Hz, J2 = 4 Hz); 13C NMR (100 MHz, d6-DMSO): δ 153.5, 143.3, 140.3, 137.0, 134.4, 134.1, 128.8, 126.7, 126.4, 126.3, 125.6, 124.1, 123.9, 121.8, 118.6 (one carbon is not resolved); FT-IR: ν cm−1 (KBr): 3263, 3013, 1642, 1587, 1555; mass: (LCMS) 264.0 (M + 1)+.
Compound (5)
To a stirred solution of 12 (0.12 g, 0.455 mmol) in dry CH3CN (15 mL), was added compound 11a (0.12 g, 0.577 mmol) dissolved in dry CH3CN (5 mL) and the reaction mixture was refluxed for 4 days. After completion of reaction, the crude product was purified by preparative TLC using ethyl acetate as eluent to give yellowish gummy compound 13 (0.15 g, yield: 70%). Finally, based on the procedure followed in the case of compound 1, the chloride anion in 13 was exchanged with the PF6− ion using NH4PF6 (0.15 g, 0.92 mmol) in MeOH–H2O (20 mL) to give the desired compound 5 (0.16 g, yield: 86%), mp 160 °C, [α]25D = −9.5 (c = 0.524 g/100 mL, MeOH), 1H NMR (400 MHz, CDCl3 containing two drops d6-DMSO): δ 9.91 (s, 1H), 9.31 (s, 1H), 8.79 (s, 1H), 8.69 (d, 1H, J = 8 Hz), 8.43 (d, 1H, J = 8 Hz), 8.31 (d, 1H, J = 5.60 Hz), 8.04 (d, 1H, J = 8 Hz), 7.91–7.88 (m, 3H), 7.69 (d, 1H, J = 8 Hz), 7.56–7.46 (m, 3H), 5.38 (s, 2H), 4.45–4.42 (m, 1H), 3.73 (s, 3H), 2.21–2.16 (m, 1H), 0.96 (d, 6H, J = 6.40 Hz); 13C NMR (100 MHz, CDCl3 containing two drops d6-DMSO): δ 171.5, 163.8, 152.6, 140.9, 137.8, 134.3, 133.9, 133.1, 132.5, 128.5, 127.4, 127.1, 125.9, 125.5, 125.0, 121.1, 119.7, 62.3, 58.0, 52.0, 30.6, 18.8, 17.8 (one carbon in the aromatic region is unresolved); FT-IR: ν cm−1 (KBr): 3379, 3286, 3099, 2967, 1736, 1703, 1656, 1598, 1533, 1504; HRMS (TOF MS ES+): calcd for (M − PF6)+: 435.2027, found: 435.2032.(S)-3-Methyl-2-(2-(naphthalene-1-yl)actamido)-N-(pyridine-3-yl)butanamide (14)
To a stirred solution of 1-naphthylacetic acid (0.2 g, 1.07 mmol) in dry CH2Cl2 (15 mL), was added oxalyl chloride (0.2 mL, 2 equiv.) and two drops of dry DMF and the stirring was continued for 6 h. Then solvent was evaporated off in vacuo to give the desired 1-naphthylacetyl chloride. This was directly used in the next step. 1-Naphthylacetyl chloride was dissolved in dry CH2Cl2 (10 mL) and to this solution amine 9a (0.25 g, 1.2 mmol) in dry CH2Cl2 (10 mL) containing Et3N (0.25 mL, 1.5 equiv.) was added. The reaction mixture was stirred for another 12 h. After completion of the reaction, the solvent was removed under reduced pressure, water was added to the residue, and the product was extracted with CHCl3 (25 mL × 2) and dried over anhydrous Na2SO4. Evaporation of the solvent gave the crude product, which was purified by silica gel column chromatography using petroleum ether–ethyl acetate (1:9, v/v) as eluent to give the product 14 (0.25 g, yield: 64%), mp 196 °C, [α]25D = −1.99 (c = 0.502 g/100 mL, CHCl3), 1H NMR (400 MHz, CDCl3): δ 9.05 (s, 1H), 8.55 (s, 1H), 8.30 (d, 1H, J = 4 Hz), 7.91 (d, 1H, J = 8 Hz), 7.86 (dd, 2H, J1 = 8 Hz, J2 = 2 Hz), 7.81 (d, 1H, J = 8 Hz), 7.50–7.39 (m, 4H), 7.15 (dd, 1H, J1 = 8 Hz, J2 = 4 Hz), 6.22 (d, 1H, J = 8 Hz), 4.47 (t, 1H, J = 8 Hz), 4.08 (dd, 2H J1 = 28 Hz, J2 = 16 Hz), 1.98–1.93 (m, 1H), 0.80 (d, 3H, J = 6.80 Hz), 0.62 (d, 3H, J = 6.80 Hz); 13C NMR (100 MHz, CDCl3): δ 171.9, 170.6, 145.0, 141.5, 134.7, 133.8, 131.8, 130.5, 128.8, 128.5, 128.3, 127.1, 126.6, 126.0, 125.6, 123.5, 123.4, 59.3, 41.3, 30.8, 19.1, 18.1; FT-IR: ν cm−1 (KBr): 3257, 3047, 2959, 1660, 1637, 1534; mass: (LCMS) 362.0 (M + 1)+.
Compound (6)
To a stirred solution of 14 (0.15 g, 0.415 mmol) in dry CH3CN (20 mL), was added compound 11a (0.11 g, 0.529 mmol) in dry CH3CN (5 mL) and the reaction mixture was refluxed for 3 days. The crude product was purified by preparative TLC using ethyl acetate as eluent to give yellowish gummy compound 15 (0.147 g, yield: 62%). Finally, according to the ion exchange procedure followed for the synthesis of 1, the chloride anion in 15 was exchanged with the PF6− ion using NH4PF6 (0.14 g, 0.86 mmol) in MeOH–H2O to give the desired compound 6 (0.15 g, yield: 85%), [α]25D = 16.33 (c = 0.612 g/100 mL, CH3CN), 1H NMR (400 MHz, CDCl3 containing two drops of d6-DMSO): δ 10.68 (s, 1H), 9.19 (s, 1H), 8.41 (d, 1H, J = 8 Hz), 8.30 (t, 2H, J = 8 Hz), 7.99 (d, 1H, J = 8 Hz), 7.86–7.84 (m, 1H), 7.80 (d, 1H, J = 8 Hz), 7.75–7.71 (m, 1H), 7.50–7.44 (m, 4H), 6.86 (d, 1H, J = 8 Hz), 5.33 (s, 2H), 4.41–4.35 (m, 2H), 4.11 (d, 2H, J = 16 Hz), 3.73 (s, 3H), 2.21–2.16 (m, 1H), 1.99–1.97 (m, 1H), 0.96–0.94 (m, 6H), 0.80 (d, 3H, J = 6.80 Hz), 0.71 (d, 3H, J = 6.80 Hz); 13C NMR (100 MHz, CDCl3 containing two drops of d6-DMSO): δ 171.9, 171.7, 171.6, 163.9, 140.0, 138.9, 135.5, 134.5, 133.5, 131.9, 131.3, 128.5, 128.0, 127.9, 127.4, 126.3, 125.8, 125.5, 123.8, 62.3, 59.4, 58.2, 52.1, 30.8, 30.57, 30.50, 19.1, 18.8, 17.9, 17.8; FT-IR: ν cm−1 (KBr): 3631, 3406, 3103, 2968, 1740, 1693, 1656, 1596, 1537, 1508; HRMS (TOF MS ES+): calcd for (M − PF6)+: 533.2758, found: 533.2750.Preparation of anionic guests
All the tetrabutylammonium salts were prepared by adding 1 equivalent of tetrabutylammonium hydroxide in methanol to a solution of carboxylic acid (1 equivalent) in methanol. The mixture was stirred at room temperature for 3 h and evaporated to dryness under reduced pressure. The gummy mass was further dried under vacuum for 24 h.Binding constant determination14
Binding constant values were determined using the fluorescence titration data. The nonlinear fit of the titration data was done using equation: I = I0 + (Ilim − I0)/2CH{CH + CG + 1/Ka − [(CH + CG + 1/Ka)2 − 4CHCG]1/2} where I represents the intensity; I0 represents the intensity of pure host; CH and CG are the corresponding concentrations of host and anionic guest; Ka is the binding constant. The binding constant (Ka) and the correlation coefficient (R) were obtained from a nonlinear least-square analysis of I vs. CH and CG.Quantum yield determination
Quantum yields of the compounds were determined in CH3CN by the relative comparison procedure using naphthalene as standard (ΦNap = 0.23 in cyclohexane).12a The general equation used in the determination of relative quantum yields is as follows:12b–c| Φu = (Φs × Fu × As × λexs × ηu2)/(Fs × Au × λexu × ηs2) |
Acknowledgements
We thank DST New Delhi, India, for financial support [project SR/S1/OC-76/2010(G); Date: 23.08.2011]. A.M. thanks CSIR, New Delhi, India for a fellowship.References
- (a) T. D. James, Top. Curr. Chem., 2005, 255, 31 Search PubMed; (b) J. Lin, Q. S. Hu, M. H. Xu and L. Pu, J. Am. Chem. Soc., 2002, 124, 2088 CrossRef CAS PubMed; (c) Z. B. Li, J. Lin, Y. C. Qin and L. Pu, Org. Lett., 2005, 7, 3441 CrossRef CAS PubMed; (d) G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723 CrossRef CAS PubMed; (e) M. Matsushita, K. Yoshida, N. Yamamoto, P. Wirscing, R. A. Lerner and K. D. Janda, Angew. Chem., Int. Ed., 2003, 42, 5984 CrossRef CAS PubMed; (f) P. Lustenberger, E. Martinborough, T. M. Denti and F. Diederich, J. Chem. Soc., Perkin Trans. 2, 1998, 747 RSC; (g) W. S. Weiner and A. D. Hamilton, Bioorg. Med. Chem. Lett., 1998, 8, 681 CrossRef CAS.
- (a) L. Pu, Chem. Rev., 2004, 104, 1687 CrossRef CAS PubMed; (b) G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1 CrossRef CAS PubMed.
- (a) G. A. Korbel, G. Lalic and M. D. Shair, J. Am. Chem. Soc., 2001, 123, 361 CrossRef CAS; (b) G. T. Copeland and S. J. Miller, J. Am. Chem. Soc., 1999, 121, 4306 CrossRef CAS; (c) K. D. Shimizu, M. L. Snapper and A. H. Hoveyda, Chem.–Eur. J., 1998, 4, 1885 CrossRef CAS.
- Fluorescent receptors for chiral amines: (a) T. Kida, T. Iwamoto, H. Asahara, T. Hinoue and M. Akashi, J. Am. Chem. Soc., 2013, 135, 3371 CrossRef CAS PubMed. Fluorescent receptors for chiral amino alcohols: (b) X. He, Q. Zhang, X. H. Liu, L. L. Lin and X. M. Feng, Chem. Commun., 2011, 47, 11641 RSC; (c) J. Jiao, X. Liu, X. Mao, J. Li, Y. Cheng and C. Zhu, New J. Chem., 2013, 37, 317 RSC. Fluorescent receptors for chiral acids: (d) Z. B. Li, J. Lin, M. Sabat, M. Hyacinth and L. Pu, J. Org. Chem., 2007, 72, 4905 CrossRef CAS PubMed. Fluorescent receptors for chiral amino acids: (e) H. Qin, Y. He, C. Hu, Z. Chen and L. Hu, Tetrahedron: Asymmetry, 2007, 18, 1769 CrossRef CAS PubMed; (f) M. H. Rodriguez and E. Juaristi, Tetrahedron, 2007, 63, 7673 CrossRef PubMed; (g) Z. B. Li, J. Lin, H. C. Zhang, M. Sabat, M. Hyacinth and L. Pu, J. Org. Chem., 2004, 69, 6284 CrossRef CAS PubMed; (h) X. Wu, M. Xie, X. Zhao, X. Liu, L. Lin and X. Feng, Tetrahedron Lett., 2014, 55, 3446 CrossRef CAS PubMed. Fluorescent receptors for carbohydrates: (i) A. Berthod, X. Chen, J. P. Kullman and D. W. Armstrong, Anal. Chem., 2000, 72, 1767 CrossRef CAS; (j) P. Bako, G. Keglevich, Z. Rapi and L. Tke, Curr. Org. Chem., 2012, 16, 297 CrossRef CAS.
- (a) V. D. Jadhav and F. P. Schmidtchen, Org. Lett., 2006, 8, 2329 CrossRef CAS PubMed; (b) A. Bencini, C. Coluccini, A. Garau, C. Giorgi, V. Lippolis, L. Messori, D. Pasini and S. Puccioni, Chem. Commun., 2012, 48, 10428 RSC; (c) F. Han, L. Chi, X. Liang, S. Ji, S. Liu, F. Zhou, Y. Wu, K. Han, J. Zhao and T. D. James, J. Org. Chem., 2009, 74, 1333 CrossRef CAS PubMed; (d) K. X. Xu, X. J. Wu, Y. B. He, S. Y. Liu, G. Y. Qing and L. Z. Meng, Tetrahedron: Asymmetry, 2005, 16, 833 CrossRef CAS PubMed; (e) J. Lin, H. C. Zhang and L. Pu, Org. Lett., 2002, 4, 3297 CrossRef CAS PubMed; (f) L. Chi, J. Zhao and T. D. James, J. Org. Chem., 2008, 73, 4684 CrossRef CAS PubMed; (g) F. Song, G. Wei, L. Wang, J. Jiao, Y. Cheng and C. Zhu, J. Org. Chem., 2012, 77, 4759 CrossRef CAS PubMed and references cited therein.
- (a) R. Datta, S. P. Tsai, P. Bonsignore, S. H. Moon and J. R. Frank, FEMS Microbiol. Rev., 1995, 16, 221 CrossRef CAS PubMed; (b) Y. J. Wee, J.-N. Kim and H. W. Ryu, Food Technol. Biotechnol., 2006, 44, 163 CAS.
- (a) M. Almaraz, C. Raposo, M. Martin, M. C. Caballero and J. R. Moran, J. Am. Chem. Soc., 1998, 120, 3516 CrossRef CAS; (b) A. Barnard, S. J. Dickson, M. J. Peterson, A. M. Todd and J. W. Steed, Org. Biomol. Chem., 2009, 7, 1554 RSC; (c) G. K. Budnikov, G. A. Evtyugin, Y. G. Budnikova and V. A. Alfonsov, J. Anal. Chem., 2008, 63, 2 CrossRef CAS; (d) L. Zhu and E. V. Anslyn, J. Am. Chem. Soc., 2004, 126, 3676 CrossRef CAS PubMed; (e) N. Seurre, K. L. Barbu-Debus, F. Lahmani, A. Zehnacker, N. Borho and M. A. Suhm, Phys. Chem. Chem. Phys., 2006, 8, 1007 RSC; (f) T. Ema, D. Tanida, K. Hamada and T. Sakai, J. Org. Chem., 2008, 73, 9129 CrossRef CAS PubMed.
- K. Ghosh and A. Majumdar, Tetrahedron Lett., 2013, 54, 5686 CrossRef CAS PubMed.
- L. Salem, X. Chapuisat, G. Segal, P. C. Hiberty, C. Minot, C. Leforestier and P. Sautet, J. Am. Chem. Soc., 1987, 109, 2887 CrossRef CAS.
- K. Ghosh, A. R. Sarkar, T. Sarkar, S. Panja and D. Kar, RSC Adv., 2014, 4, 20114 RSC.
- M. T. Albelda, E. Garcia-Espana, L. Gil, J. C. Lima, C. Lodeiro, J. S. de Malo, M. J. Melo, A. J. Parola, F. Pina and C. Soriano, J. Phys. Chem. B, 2003, 107, 6573 CrossRef CAS.
- (a) A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213 CrossRef CAS; (b) R. A. Velapoldi and H. H. Tonnesen, J. Fluoresc., 2004, 14, 465 CrossRef CAS; (c) Y. Wu, X. Peng, J. Fan, S. Gao, M. Tian, J. Zhao and S. Sun, J. Org. Chem., 2007, 72, 62 CrossRef CAS PubMed.
- P. Job, Ann. Chim., 1928, 9, 113 CAS.
- B. Valeur, J. Pouget, J. Bourson, M. Kaschke and N. P. Eensting, J. Phys. Chem., 1992, 96, 6545 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.2), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures showing the change in fluorescence and UV-vis titrations of receptors 1–6 with various chiral anions, Job plot, binding constant table, 1H NMR titration and other spectral data. See DOI: 10.1039/c5ra00017c |
| This journal is © The Royal Society of Chemistry 2015 |