
A polyoxometalate-assisted approach for synthesis of Pd nanoparticles on graphene nanosheets: synergistic behaviour for enhanced electrocatalytic activity†
Abstract
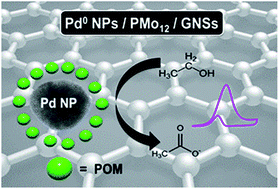
A polyoxometalate (POM) assisted approach has been employed to prepare a nanohybrid of Pd nanoparticles (PdNPs) and graphene nanosheets (GNSs). The Keggin-type POM, phosphomolybdic acid (PMo12), was applied to serve as both reducing and stabilising agent. The as-prepared nanohybrid (Pd/PMo12/GNSs) was comprehensively characterised using transmission electron microscopy and X-ray diffraction analysis. The synergistic behaviour of PdNPs, PMo12 and GNSs in the nanohybrid leads to elevated electrocatalytic property for ethanol oxidation. Moreover, the Pd/PMo12/GNSs nanohybrid was activated by applying a sufficiently negative potential which plays a key role in promoting the electrocatalytic activity. The activated catalyst presents a superior performance towards ethanol electrooxidation reaction and shows better tolerance to poisoning species compared to Pd and Pt nanoparticles. The outstanding electrocatalytic activity of the tri-component (Pd/PMo12/GNSs) nanohybrid is discussed with relevance to its application in direct alcohol fuel cells (DAFCs).
 Please wait while we load your content...
Please wait while we load your content...

