DOI:
10.1039/C5RA00969C
(Paper)
RSC Adv., 2015,
5, 21831-21842
Diphenylether based derivatives as Fe(III) chemosensors: spectrofluorimetry, electrochemical and theoretical studies†
Received
16th January 2015
, Accepted 18th February 2015
First published on 18th February 2015
Abstract
4-(2,4-Dinitrophenoxy)-3-methoxybenzaldehyde (DPE-I) and (4-(2,4-dinitrophenoxy)-3-methoxyphenyl)methanol (DPE-II) were synthesized with high yield and characterized by spectroscopic techniques. The complexation behaviours of DPE-I and DPE-II for various cationic species (Na+, K+, Mg2+, Cu2+, Ba2+, Cr3+, Fe3+, Co2+, Ni2+, Zn2+, Cd2+, and Pb2+) in HEPES buffered CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O system were investigated and compared by spectroscopic and voltammetric techniques. Both receptors showed high degree of selectivity for Fe3+ over other cations. Receptors showed 1:1 complexation with Fe3+. DPE-I showed fluorescence quenching on complexation with Fe3+ ion at 350 nm due to PET (photon induced transfer) mechanism between Fe3+ and the large π electron conjugate system of ligand, while DPE-II displayed emission band at 314 nm which underwent fluorescence quenching selectively on gradual addition of Fe3+ at 314 nm and simultaneously a new band at 424 nm emerged with isosbestic point at 355 nm which increases with increase in Fe3+ conc. Electrochemically, DPE-I due to presence of electron withdrawing (aldehydic group) facilitates reduction of nitro group on benzene ring of the molecule so, peak potentials of the two curves obtained for DPE-I appear at −1.16 V (Epc1) and −1.39 V (Epc2) respectively, while in DPE-II (methanolic group) due to its electron donating character makes the reduction of nitro group a little difficult due to which peak potentials occur at higher potential as compared to DPE-I i.e. at −1.30 V (Epc1) and −1.43 V (Epc2) respectively.
:H2O system were investigated and compared by spectroscopic and voltammetric techniques. Both receptors showed high degree of selectivity for Fe3+ over other cations. Receptors showed 1:1 complexation with Fe3+. DPE-I showed fluorescence quenching on complexation with Fe3+ ion at 350 nm due to PET (photon induced transfer) mechanism between Fe3+ and the large π electron conjugate system of ligand, while DPE-II displayed emission band at 314 nm which underwent fluorescence quenching selectively on gradual addition of Fe3+ at 314 nm and simultaneously a new band at 424 nm emerged with isosbestic point at 355 nm which increases with increase in Fe3+ conc. Electrochemically, DPE-I due to presence of electron withdrawing (aldehydic group) facilitates reduction of nitro group on benzene ring of the molecule so, peak potentials of the two curves obtained for DPE-I appear at −1.16 V (Epc1) and −1.39 V (Epc2) respectively, while in DPE-II (methanolic group) due to its electron donating character makes the reduction of nitro group a little difficult due to which peak potentials occur at higher potential as compared to DPE-I i.e. at −1.30 V (Epc1) and −1.43 V (Epc2) respectively.
Introduction
Synthesizing sensitive and selective chemosensors for specific metal detection is an interesting topic due to challenges in molecular design and their applications in a number of research fields.1–4 Ionophores with functional groups like carboxylate,5,6 sulfonate,7,8 phosphonate,9–11 carbohydrate12,13 and ammonium14–16 can selectively trap metals ions. However, none of the attempts has been made to use diphenylether based receptor having (R–O–R) ethereal linkage for metal detection. This is our first report using diphenylether based receptors for detection of iron.
Iron plays a significant role in many biochemical processes17,18 and is an important metal ion for most organisms. Fe3+ acts as a cofactor in many enzymatic reactions involved in the mitochondrial respiratory chain, and both its deficiency and excess can induce a variety of diseases.18–20 A surplus of iron induces oxidative damage, rendering the intracellular scavenging of iron a major therapeutic target.21 Therefore, methods of ferric ion detection have received a great deal of study. However, in spite of the important role of Fe3+ in chemical and biological processes, selective and sensitive chemosensors for Fe3+ are still rare.
In the past few years, several analytical techniques for the determination of trace amounts of Fe3+ have been reported, including atomic absorption spectrometry (AAS),22 inductively coupled plasma-atomic emission spectrometry (ICP-AES),23 inductively coupled plasma-mass spectrometry (ICP-MS),24 electrochemical methods25,26 etc. Although these methods offer good limits of detection and wide linear ranges, most of these require the use of expensive and sophisticated instrumentation and complicated pretreatment procedures, not inappropriate for on-line or in-field monitoring. Owing to their advantages of simple equipment, rapid response, high sensitivity and easy operation, fluorescent approaches have been developed for the determination of Fe3+. Currently, there have been a lot of developments in the field of Fe3+ chemosensors which have been used as fluorescent probes.27–41 A comparative study of the chemosensors for Fe3+ has been shown in Table S1.†42,7,3,43,44 However, many of these probes exhibit either one or two of the following features that limit their practical applications. Firstly, most of the reported Fe3+ probes cannot work in aqueous media due to their hydrophobicity and low binding affinity. Secondly, most of these have interference problems caused by other transition metal cations such as Cu2+, Co2+, Al3+ or Hg2+. Therefore, searching for highly selective chemosensors for Fe3+ in aqueous solutions is of great importance.
With purely organic hosts, appropriate geometry can sometimes be a problem for a proper host–guest interaction. On the other hand, metal ions can profitably orient the ligands in right conformations so that their hydrogen bond donor groups can converge towards an external guest. So, in this report, we have developed widespread diphenylether based chemosensors which get oriented in proper conformation to best fit the Fe3+ and hence can be used in various fields of analysis, as it can be used as a fluorescent chemosensor as well as electrochemical chemosensor. In this manuscript, complexation behavior of 4-(2,4-dinitrophenoxy)-3-methoxybenzaldehyde DPE-1 and (4-(2,4-dinitrophenoxy)-3-methoxyphenyl)methanol DPE-II with various cations (Na+, K+, Cr3+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Ba2+, Hg2+, Cd2+ and Pb2+) were investigated in aqueous media by spectrophotometric and voltammetric methods and the binding mode was further studied by using Gauss View 4.1.2 software.
Material and instrumental methods
Reagents and measurements
All reagents were purchased from commercial suppliers and used without further purification. Acetonitrile was distilled and was stored on molecular sieves before use. TLC analyses were performed on silica gel plates and column chromatography was conducted over silica gel (mesh 100–120). 1H NMR spectra were recorded using JEOL A1 spectrometer operating at 400 MHz. 13C NMR spectra was recorded at 100 MHz. All chemical shifts are reported in ppm relative to the TMS as an internal reference. UV-Vis studies were carried out on a Analytic Jena machine using slit width of 1.0 cm and matched quartz cells. Fluorescence spectra were determined on a Perkin Elmer LS-55 fluorescence spectrometer.
Electrochemical measurements were carried out on Gamry Potentiostat/Galvanostat/ZRA Interface 1000. Solvents (acetonitrile, dichloromethane, dimethyl formamide) used in the spectrophotometric and electrochemical experiments were of HPLC grade (Sd Fine, India) and used as obtained. For cation interaction investigation, nitrate (Loba Chemie, India) and perchlorate salts (Sigma Aldrich) of the metal were used. Tetrabutylammonium hexafluorophosphate (Sigma Aldrich) was used as a supporting electrolyte in all the voltammetric experiments. The working electrode was a glassy carbon electrode (CH Instruments, USA, 2 mm diameter). Platinum electrode was used as a counter electrode. All the potentials were reported against Ag/Ag+ reference electrode. The Ag/Ag+ electrode contained an internal solution of 0.01 M AgNO3 and 0.1 M TBAPF6 in solvents like acetonitrile, dimethyl formamide and dichloromethane. The working GC electrode was polished with alumina followed by washing with water and solvent before each cyclic voltammogram. The electrochemical measurements were carried out at a temperature of 25.0 ± 1 °C. In all experiments, the test solutions were de-aerated by a stream of N2 gas purging through the solution for at least 4–5 minutes.
Further, theoretical studies were performed by using Gauss View 4.1.2 software to know the binding mode of selective metal ion. For metal ion titrations, the solutions were prepared in CH3CN:H2O (9:1, v/v) containing 10 mM HEPES buffer. Stock solution of DPE-I and DPE-II (10−3 M) were prepared in CH3CN.
Synthesis
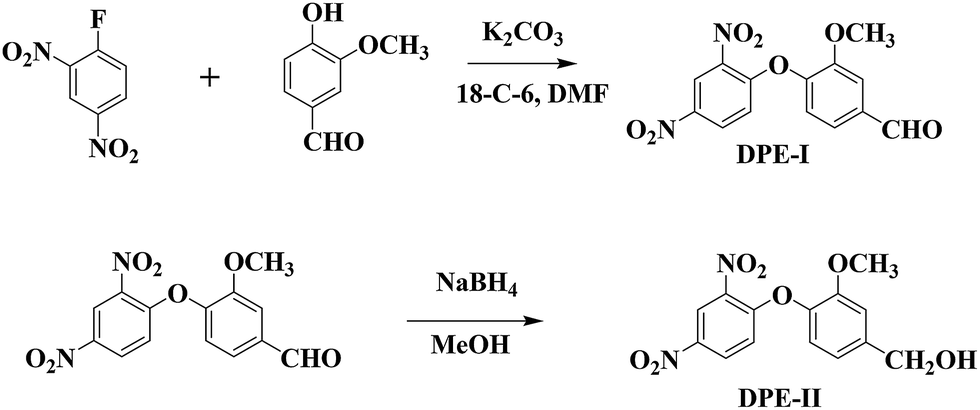
Compounds were synthesized as shown in Scheme 1 according to methods described in the literature.45 Synthesized compounds were confirmed for their purity by melting point, TLC, 1H and 13C NMR spectroscopy which was in accordance with the previously reported literature (Fig. S1 and S2†).
 |
| | Scheme 1 Synthesis procedure of DPE-I and DPE-II compounds. | |
Preparation of metal–ligand complexes
The binding ability studies of DPE-I and DPE-II were carried out with different metal ions viz. Na+, K+, Cr3+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Ba2+, Hg2+, Cd2+ and Pb2+ using spectroscopic and voltammetric experiments. DPE-I and DPE-II were taken in ACN:H2O (9:1) medium. The solvent medium was prepared by taking 10 mM of HEPES buffer to maintain pH = 7. Ligands DPE-I and DPE-II were taken in 20 μM amounts to which 50 equivalents of each metal was added separately to prepare the metal–ligand complex solutions at 25 °C. Those solutions of each metal with ligands were subjected to different studies on spectrophotometry, spectrofluorimetry and voltammetry. Each study was conducted within 5 minutes of preparation of each metal–ligand complex.
Results and discussion
UV-Vis studies
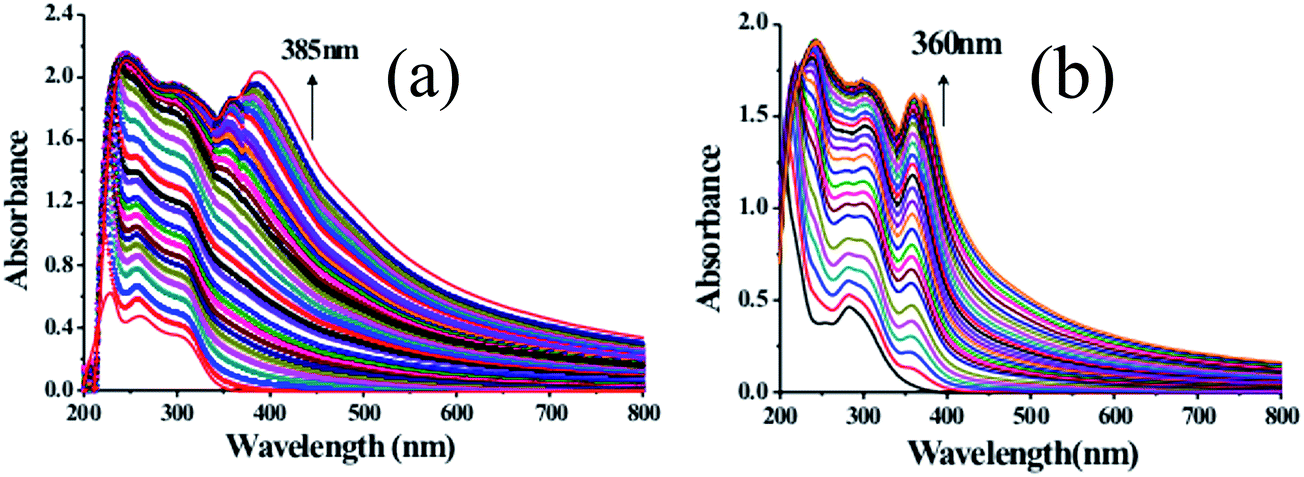
The UV-Vis spectroscopic studies of receptors DPE-I and DPE-II (20 μM) were investigated in HEPES buffered CH3CN:H2O (9:1; v/v) solvent system at pH 7.0 ± 0.1 because the pure CH3CN solvent developed turbidity with receptors. DPE-I showed an absorption band at 240 nm while DPE-II showed an absorption band at 280 nm due to n → π* transition. On the addition of different metal ions, viz. Na+, K+, Cr3+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Ba2+, Hg2+, Cd2+ and Pb2+ to solutions of DPE-I and DPE-II, no significant changes in their respective UV-Vis spectra were observed except in the case of Fe3+ ions as shown in Fig. S3.† In order to evaluate the complete structural behaviour of DPE-I and DPE-II towards Fe3+, titrations of DPE-I and DPE-II were performed with different concentrations of the metal ion. On the addition of 17 equivalents of Fe3+ to the solution of DPE-I (Fig. 1a), the absorption band at 240 nm gets flattened. A new band appears at 385 nm at 18 equivalents of Fe3+. On further addition of iron solution there was an increase in absorption intensity at 385 nm which attains a saturation point after 46 equivalents. Similarly, for DPE-II, the absorption band at 280 nm gets flattened on the addition of 11 equivalents of Fe3+, and a new band appears at 360 nm at 14.5 equivalents. On further addition of iron solution there was an increase in the absorption intensity of the band at 360 nm which attains a saturation point after 26.5 equivalents of Fe3+ as shown in Fig. 1b. The absorption ratio at (240 nm/385 nm) two wavelengths varied from 0.03987 to 1.34262, indicating 33.67 fold absorption ratio changes for DPE-I while for DPE-II absorption ratio at two wavelengths (280 nm/360 nm) varied from 0.07597 to 0.96272, indicating 12.67 fold absorption ratio changes. Appearance of new bands at 385 nm and 360 nm, respectively for DPE-I and DPE-II on addition of Fe3+ (17 equivalents) indicate formation of the corresponding complexes. Further addition of Fe3+ ions resulting in increase in absorbance is due to the increase in concentration of the resulting metal–ligand complexes.
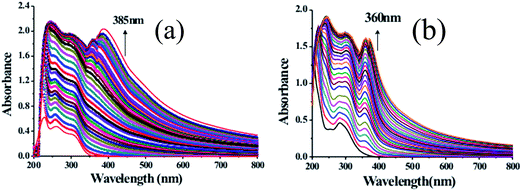 |
| | Fig. 1 Absorption spectra of (a) DPE-I (20 μM) and (b) DPE-II (20 μM) at pH 7.0 ± 0.1 (10 mM HEPES in CH3CN:H2O 9:1, v/v) upon complexation with increasing concentration of Fe3+ ions. | |
Thus, these chemo sensors can be used to estimate a wide range of Fe3+ ions ratio metrically between concentrations of 2 × 10−5 to 1 × 10−3 M and 2 × 10−5 to 6 × 10−4 M for DPE-I and DPE-II respectively, as shown in Fig. 2a and b. The lower detection limit and stability constants for the complexes are shown in Table 1.
 |
| | Fig. 2 Ratiometric response of different concentrations of Fe3+ ions on (a) DPE-I (20 μM) and (b) DPE-II (20 μM) at pH 7.0 ± 0.1 (10 mM HEPES in CH3CN:H2O 9:1, v/v). | |
Table 1 Stability constants and some parameters of the DPE-I and DPE-II in the presence of Fe3+ in HEPES buffered CH3CN:H2O system
| Complex |
Coefficient of regression (R2) |
Stability constant |
Detection limit |
| DPE-I + Fe3+ |
0.999 |
1.3 × 103 M−1 |
4.23 × 10−4 M |
| DPE-II + Fe3+ |
0.998 |
3.0 × 103 M−1 |
2.93 × 10−4 M |
To study practical applicability of DPE-I and DPE-II, competitive experiments were carried out in the presence of 10 equivalents of Fe3+ ions mixed with 50 equivalents of each of the different metal ions. No significant variations in the absorption spectra were observed in the presence of a number of interfering metal ions like Na+, K+, Cr3+, Fe3+, Ni2+, Cu2+, Zn2+, Ba2+, and Pb2+. Similar behaviour was observed for DPE-I and DPE-II compounds (Fig. 3a and b).
 |
| | Fig. 3 Black bars represent selectivity of (a) DPE-I and (b) DPE-II upon addition of different metal ions at pH 7.0 ± 0.1 (10 mM HEPES in CH3CN:H2O 9:1, v/v) and red bars show the competitive selectivity of ligands in the presence of interfering metal ions. | |
Emission properties of receptors
To obtain an insight into the fluorescent properties of the receptor, DPE-I was excited at 240 nm and displayed emission band at 350 nm. Further, complexation with different metal ions such as Na+, K+, Cr3+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Ba2+, Hg2+, Cd2+ and Pb2+ in CH3CN:H20 (9:1, v/v) were studied, which underwent fluorescence quenching selectively on addition of Fe3+, whereas other metal ions did not show any significant change. While DPE-II was excited at 280 nm which displayed emission band at 314 nm which underwent fluorescence quenching selectively on gradual addition of Fe3+. Simultaneous to the quenching process a new band at 424 nm emerged with isosbestic point at 355 nm. The emission at 424 nm increases with increase in Fe3+ conc., whereas other metal ions did not show any significant change. In order to evaluate the complete structural behaviour of DPE-I and DPE-II towards Fe3+, the fluorescence titrations of DPE-I and DPE-II were performed at different concentrations of metal ions as shown in Fig. 4a and b.
 |
| | Fig. 4 (a) Fluorescence spectra of (a) DPE-I and (b) DPE-II (20 μM) at pH 7.0 ± 0.1 (10 mM HEPES in CH3CN:H2O 9:1, v/v), upon complexation with increasing concentration of Fe3+ ions. | |
Ratiometric behaviour of DPE-I and DPE-II in presence of different Fe3+ concentrations is shown in Fig. S4.† In the fluorescence titration of DPE-I with Fe3+ ions, the plot of I0/I (I0 and I, fluorescence intensity of DPE-I without and with Fe3+ ions, respectively) vs. Fe(III) concentration was not a straight line. This indicated that the fluorescence quenching of DPE-I by Fe3+ ions did not obey the Stern–Volmer quenching equation (I0/I = 1 + K[Fe]). Therefore, the fluorescence quenching is a static quenching. The disappearance in fluorescence intensity of DPE-I on interaction with Fe3+ may be explained by the favourable photo induced electron transfer (PET) mechanism between ligand and Fe3+,46 while such mechanism was not supported for DPE-II. PET mechanism is observed in DPE-I as the lone pairs on oxygen of –CHO are engaged through conjugation with Fe3+. In case of DPE-II, the lone pairs on oxygen of –OH not being conjugated with the benzene ring are available for excitation resulting in emission band at 424 nm and no PET mechanism is observed.
The linear ratiometric response (I424/I314) in case of DPE-II, with increasing concentration of Fe3+ also makes it a unique probe for it. Presence of isosbestic point at 355 nm proves that one intermediate is formed before complete complexation which is also highly fluorescent and fluorescent intensity at 424 nm goes on increasing upto 50 equivalents of Fe3+ ions due to complex formed between DPE-II and Fe3+. The Fe3+ ions are likely to bind to the ligands via O-atoms of the nitro and methoxy groups. The PET process occurs due to the transfer of electron density from ligand to the metal ion. The higher the charge density a cation possesses (the charge density of Fe3+ is 6.09),47 the stronger the electronic affinity it provides. So, it is reasonable to be used to achieve PET processes between Fe3+ and the large π electron conjugate system of ligand. Moreover, from theoretical studies it is observed that the DPE-I is a good electron donor, which displays an ability to coordinate Fe3+ ions, so that there can be a reasonable charge transfer between DPE-I and the Fe3+ ions.
The non-linear regression analysis of fluorescence data for Fe3+–DPE-I complex showed the formation of 1:1 complex with association constant 5.1 × 103 M−1 while DPE-II showed better association constant of 7 × 103 M−1. To elicit the interactions between Fe3+ and DPE-I and DPE-II respectively, the binding stoichiometry was determined by the Job's plot.48,49 The Job's function FJob is calculated according to the equation FJob = (1 − X)F0 − F. The plot of the fluorescence versus the mole fraction of the added Fe3+ (Fig. 5a and b) shows two parts that can be fitted by straight lines. The lines intersect at X equal to 0.5. These results reveal the formation of a 1:1 complex formulated as DPE-I–Fe3+ and DPE-II–Fe3+, respectively.
 |
| | Fig. 5 Job's plot of (a) DPE-I and Fe3+ (b) DPE-II and Fe3+ which indicated the stoichiometry of L–Fe3+ complex is 1:1. | |
Electrochemical measurements
Electrochemical properties of DPE-I and DPE-II were investigated by CV and DPV. The cyclic voltammograms for DPE-I and DPE-II in CH3CN:H2O (9:1, v/v) solution containing 0.1 M TBAPF6 as the supporting electrolyte are shown in Fig. 6a and b.
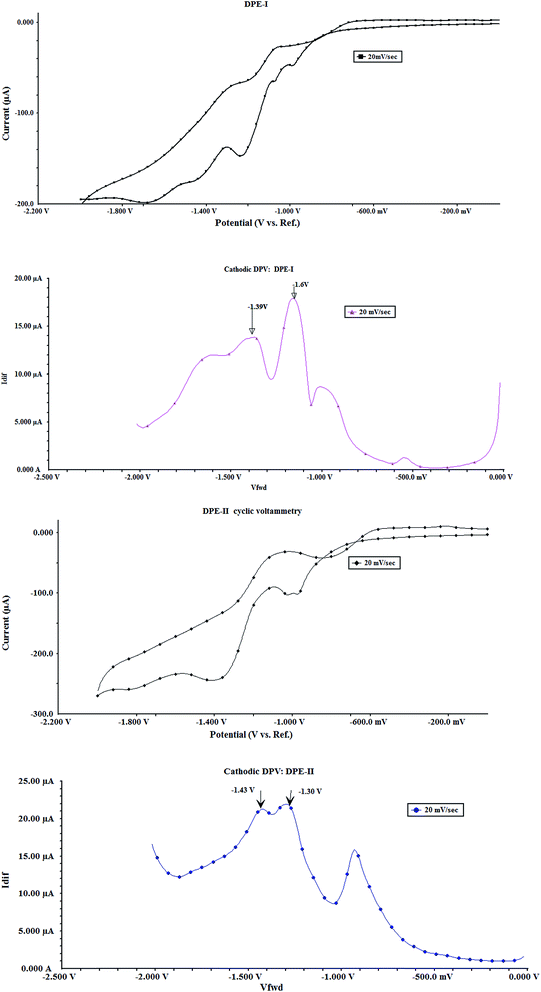 |
| | Fig. 6 Cyclic and differential pulse voltammograms of (a) DPE-I (b) DPE-II (5 × 10−4 M) at pH 7.0 ± 0.1 (10 mM HEPES in CH3CN:H2O 9:1, v/v) using 0.1 M TBAPF6 as supporting electrolyte, scan rate 20 mV s−1 and GC as working electrode. Modulation amplitude for DPV: 50 mV s−1. | |
Electrochemical behaviour of DPE-I and DPE-II
Synthesis of DPE-I and DPE-II containing electron donating and electron withdrawing substituents, respectively was done to check their remote substituent effect on the electronic environment around the ethereal oxygen of the diphenylether group. These compounds are identified as DPE-I (containing –CHO group meta to methoxy group on benzene ring) and DPE-II (containing –CH2OH group meta to methoxy group on benzene ring). It is hypothicated that the aldehydic group should facilitate reduction of nitro group on benzene ring of the molecule, while methanolic group, due to its electron donating character, should make the reduction of nitro group, a little difficult. Peak potentials of the two curves obtained for DPE-I appear at −1.16 V (Epc1) and −1.39 V (Epc2), respectively, whereas corresponding peaks for DPE-II are observed at −1.30 V (Epc1) and −1.43 V (Epc2), respectively (Fig. 6a and b). Corresponding peak currents for the respective peak potentials are shown in Table 2.
Table 2 Peak potential (V) and peak current (μA) values for DPE-I and DPE-II (5 × 10−4 M), scan rate 100 mV s−1
| Compound |
Peak potentials (V) |
Cathodic peak currents (μA) |
| Epc1 |
Epc2 |
ipc1 |
ipc2 |
| DPE-I |
−1.16 |
−1.39 |
−45.7 |
−37.4 |
| DPE-II |
−1.30 |
−1.43 |
−62.6 |
−76.5 |
Effect of scan rate
In order to understand nature of the electrode processes taking place for DPE-I and DPE-II, peak currents of cathodic waves were plotted against square root of scan rate (Fig. 7). A linear relation between peak current and square root of scan rate shows that Fick's law of diffusion is obeyed and the charge transport is diffusion based process. For DPE-II with increase in scan rate from 20 mV s−1 to 100 mV s−1, two separate reduction peaks at −1.30 V and −1.43 V merge to form a single broad reduction peak. It indicated that at higher scan rates the two different reduction processes were observed as a single step, while for DPE-I both the reduction steps were observed as separate peaks even at 200 mV s−1 or higher scan rates. This observation is also supported by the DPV curves (Fig. S5†).
 |
| | Fig. 7 Plots of peak current and square root of scan rate for DPE-I and DPE-II. | |
Effect of concentration of the electroactive compound
Electrochemical study of the compounds DPE-I and DPE-II was done to know the effect of different concentrations on their respective CV behaviors. Results are shown in Fig. S6.† With increase in concentration of each of DPE-I and DPE-II, a corresponding increase in the respective peak currents was observed along with a cathodic shift in the peak potential. This is due to the reason that increase in concentration of redox substances makes the velocity of mass diffusion rapid and hence the electrode processes become more controlled by the electrode reactions. Calibrations for concentration of analyte species and cathodic peak currents were plotted and observed coefficients of regression varied within range 0.98–0.99, as shown in Fig. 8.
 |
| | Fig. 8 Calibration curves between concentrations of receptors and peak current for DPE-I and DPE-II. | |
Effect of solvent
Receptors DPE-I and DPE-II were studied for their voltammetry in different solvent systems like dichloromethane (DCM), acetonitrile (ACN) and N,N-dimethylformamide (DMF). In addition, mixed solvent systems of ACN:H2O (9:1, v/v) and DMF:H2O (9:1, v/v) were also taken for the study anticipating some interactions of water with substituents –CHO in DPE-I and –CH2OH in DPE-II. Cyclic voltammetric and differential pulse voltammetric graphs for DPE-I and DPE-II in different solvent media are shown in Fig. S7.†
DPV peaks for both the compounds in all solvent systems are identified in Table 3. It is interesting to note that DPE-I shows only one reduction peak while DPE-II shows three reduction peaks in ACN solvent system. These can be explained as below.
Table 3 Peak potentials (V) for DPE-I and DPE-II (5 × 10−4 M) in different solvent systems
| Solvent |
Dipole moment (D) |
DPE-I |
DPE-II |
| Epc1 (V) |
Epc2 (V) |
Epc3 (V) |
Epc1 (V) |
Epc2 (V) |
Epc3 (V) |
| ACN |
3.92 |
−1.36 |
— |
— |
−1.32 |
−1.68 |
−1.85 |
| DMF |
3.86 |
−1.25 |
−1.48 |
−1.87 |
−1.348 |
−1.70 |
−1.89 |
| DCM |
1.60 |
−1.23 |
−1.47 |
−1.750 |
−1.290 |
−1.62 |
−1.82 |
| ACN:H2O |
|
−1.16 |
−1.39 |
— |
−1.30 |
−1.43 |
— |
In receptor DPE-I there are three prominent functional groups like –CHO, –OCH3 and –NO2 which can be responsible for the cathodic current peaks. Amongst these three groups, because of its electron withdrawing inductive effect nitro is the most probable candidate for reduction and hence shows a peak at lowest value of applied potential i.e. −1.36 V. The absence of peaks in DPE-I (−1.68 V and −1.85 V) is due to replacement of carbonyl bearing functional group –CHO in DPE-I with –CH2OH in DPE-II. This change in functional group infact easies out electron withdrawing tendency of ethereal oxygen para to –CHO group, hence the absence of cathodic peak at −1.68 V. The cathodic peak at −1.68 V in DPE-II is probably due to methoxy group as the peak at −1.85 V in DPE-II disappears with change in solvent system from ACN to ACN:H2O (9:1, v/v) system due to interaction of –OCH3 group indicating its involvement with H2O through hydrogen bonding. Moreover, the absence of corresponding peak (−1.85 V) in DPE-I in both ACN and its mixture with water can also be justified due to poor reduction potential of –CHO (in DPE-I) as compared to –CH2OH (in DPE-II). The absence of Epc3 in ACN:H2O solvent system is clear indication of hydrogen bonding of CH2OH group with water as shown below:
In solvent media like DCM, the main observation as compared with ACN solvent system is with regard to shift in all peaks of DPE-I and DPE-II to lower potential values. This important shift is due to low dipole moment of DCM (1.60 D) as compared to ACN (3.92 D) and DMF (3.86 D). Hence, polarity of the solvent does influence reduction potentials of target molecules. It is further noticed that the reduction potentials of DPE-I and DPE-II are almost similar due to similar dipole moments of ACN and DMF solvents.
Metal–ligand complexation studies
Voltammetric studies of the synthesized diphenylether based compounds were performed to study their tendency to complex with transition metal ions so that these can be projected as ionophores suitable for heavy metal detection by simple voltammetric methods. DPE-I and DPE-II were studied for their tendency for complexation with Fe3+ ions. Voltammograms were drawn for both DPE-I and DPE-II with and without Fe3+ ions. Significant decrease in current magnitude of Ep1 and Ep2 from 45.7 μA to 9.7 μA and 37.4 μA to 11.0 μA respectively, for DPE-I prompted us to study complexation on a quantitative way as shown in Fig. 9. Incremental amounts of Fe3+ were added to 2.5 μmoles of DPE-I and the resulting current magnitude are given in Table 4. Similar experiments were carried out on DPE-II compound. It can be seen from Table 4 that with addition of increasing concentration of Fe3+, a linear fall in cathodic peak current is observed till metal ions ratio with the ligand is reached 1:1 (Fig. 10). Further addition of the metal ions did not bring any further change in current magnitude. Exactly similar trend in current magnitude was observed for DPE-II. Hence, DPE-I and DPE-II can be projected as ionophores for Fe3+ metal detection. Calibration curves for the detection of Fe3+ using DPE-I and DPE-II are shown in Fig. 10. The proposed method is valid for the detection of Fe3+ species in the concentration range 0–2.5 μmoles. Presence of other metal ions (Na+, K+, Cr3+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Ba2+, Hg2+, Cd2+ and Pb2+) have also been studied and results indicate selectivity for Fe3+ ions only.
 |
| | Fig. 9 Cyclic and differential pulse voltammograms of ligands (DPE-I and DPE-II) upon increment addition of metal (μM Fe3+) at pH 7.0 ± 0.1 (10 mM HEPES in CH3CN:H2O 9:1, v/v), 0.1 M TBAPF6. | |
Table 4 Peak current values for DPE-I and DPE-II with and without Fe3+ ion
| Ligand ‘L’ (DPE-I/DPE-II) |
DPE-I |
DPE-II |
| ip2 (−1.39 V) (μA) |
ip1 (−1.16 V) (μA) |
Δip2 (−1.39 V) (μA) |
Δip1 (−1.16 V) (μA) |
ip2 (−1.43 V) (μA) |
ip1 (−1.30 V) (μA) |
Δip2 (−1.43 V) (μA) |
Δip1 (−1.30 V) (μA) |
| Without metal (2.5 μmoles) |
−37.43 |
−45.67 |
— |
— |
−76.51 |
−62.59 |
— |
— |
| L + Fe3+ (0.5 μmoles) |
−32.64 |
−40.79 |
−4.79 |
−4.88 |
−53.79 |
−40.82 |
−22.72 |
−21.77 |
| L + Fe3+ (1.0 μmoles) |
−24.08 |
−30.01 |
−13.35 |
−15.66 |
−38.29 |
−33.56 |
−38.22 |
−29.03 |
| L + Fe3+ (1.5 μmoles) |
−20.05 |
−23.75 |
−17.38 |
−21.92 |
−27.77 |
−24.61 |
−48.74 |
−37.98 |
| L + Fe3+ (2.5 μmoles) |
−14.56 |
−14.85 |
−22.87 |
−31.07 |
−13.03 |
−15.99 |
−63.48 |
−46.60 |
| L + Fe3+ (2.8 μmoles) |
−11.01 |
−10.71 |
−26.42 |
−34.96 |
−13.02 |
−15.90 |
−63.49 |
−46.69 |
| L + Fe3+ (3.5 μmoles) |
−11.01 |
−9.73 |
−26.42 |
−35.94 |
−13.02 |
−15.90 |
−63.49 |
−46.69 |
 |
| | Fig. 10 Calibration curves showing response of metal (Fe3+) addition to ligands (a) DPE-I and (b) DPE-II. | |
Interference studies
Selectivity of DPE-I and DPE-II was confirmed further by interference studies with various interfering metal ions added in excess to the target metal ion. Voltammetric study was done to study the effect of the presence of other ions on the response of DPE-I and DPE-II for Fe3+ ions. Both the derivatives responded selectively to Fe3+ ions even in the presence of other metal ions (Table 5) as indicated from the quenching of the electroactive nature of ligands (DPE-I and DPE-II) i.e. large decrease in peak currents. After complexation with the metal ions, the receptors don't remain electroactive due to non-availability of lone pair of electrons on the heteroatoms.
Table 5 Peak potential (V) values for DPE-I and DPE-II alone and with different metal ions
| Metal studied |
DPE-I |
DPE-II |
| Ep1 (V) |
Ep2 (V) |
Ep1 (V) |
Ep2 (V) |
| Ligand |
−1.16 |
−1.39 |
−1.30 |
−1.43 |
| Na+ |
−1.20 |
−1.36 |
−1.31 |
−1.45 |
| K+ |
−1.14 |
−1.38 |
−1.23 |
−1.41 |
| Mg2+ |
−1.15 |
−1.39 |
−1.27 |
−1.41 |
| Cr3+ |
−1.01 |
−1.22 |
−1.21 |
−1.42 |
| Fe3+ |
— |
— |
— |
— |
| Co2+ |
−1.14 |
−1.36 |
−1.30 |
−1.42 |
| Cu2+ |
−1.16 |
−1.36 |
−1.28 |
−1.42 |
| Zn2+ |
−1.10 |
−1.35 |
−1.32 |
−1.44 |
| Ba2+ |
−1.16 |
−1.39 |
−1.29 |
−1.38 |
Theoretical studies
The computational study was carried out by using Density Functional Theory (DFT) in an attempt to better understand the nature of receptors and their interaction with metal ion. The B3LYP function was employed for calculations with 6-31G basis set on Gaussian 03W programmer except for Fe3+, for which LanL2DZ was used.50–53 The receptors DPE-1 and DPE-II have three dimensional structure forming a pseudo cavity between ethereal oxygen, nitrogen of nitro group and oxygen of methoxy group and B3LYP/6-31G basis set was employed for their optimization (Fig. 11a and b). However, DPE-I–Fe3+ and DPE-II–Fe3+ were optimized by using B3LYP/6-31G and LanL2DZ basis set for Fe3+ (Fig. 11c and d).
 |
| | Fig. 11 The DFT optimized structures of receptors DPE-I and DPE-II and their complexes with Fe3+ calculated at the B3LYP/6-31G/LanL2DZ level respectively. The red, blue, gray, and purple spheres refer to O, N, C, and Fe, respectively. | |
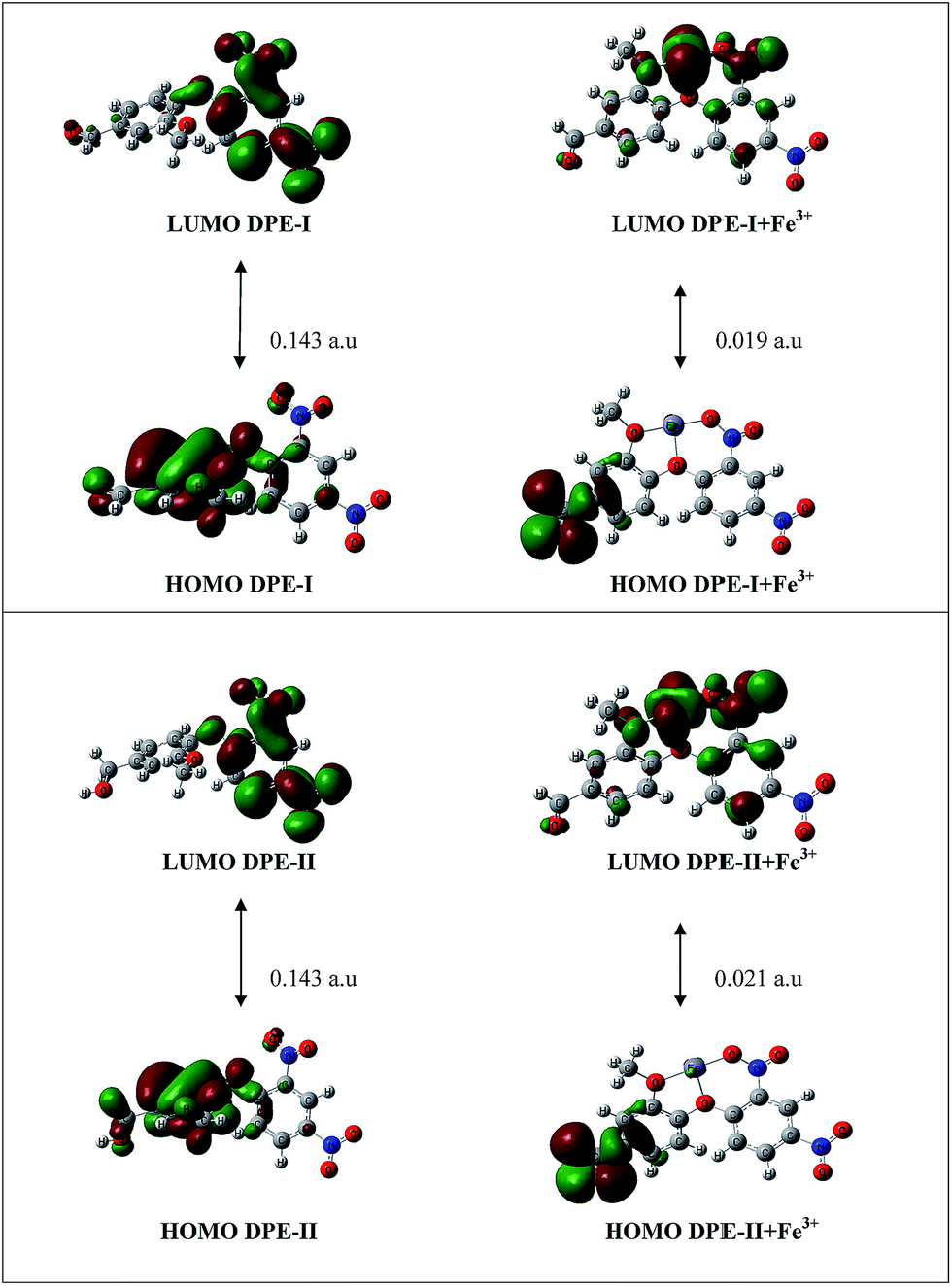
Optimized structures revealed the possible mode of complexation in which ether group, due to its flexible nature gets rotated and forms best fit metal–ligand coordination sites between Fe3+ and the receptors. On coordination with Fe3+, there is an increase in the stability of the whole system which was confirmed from the value of energy optimization (Table 6). The gap between HOMO–LUMO is less for DPE-I–Fe3+ and DPE-II–Fe3+ as compared to receptor DPE-I and DPE-II alone, as depicted in Fig. 12. Further, the HOMO–LUMO of the DPE-I + Fe3+ and DPE-II + Fe3+ showed that LUMO is main contributor in electronic transition.50,51
Table 6 Energy values (in a.u) of HOMO, LUMO and LUMO + 1 for DPE-I and DPE-II and their complexes with Fe3+
| Receptor |
Energy in a.u |
| EHOMO |
ELUMO |
ELUMO + 1 |
Egap1 |
Egap2 |
| DPE-I |
0.256 |
0.113 |
0.107 |
0.143 |
0.149 |
| DPE-I + Fe3+ |
0.487 |
0.468 |
0.435 |
0.019 |
0.052 |
| DPE-II |
0.236 |
0.093 |
0.087 |
0.143 |
0.149 |
| DPE-II + Fe3+ |
0.488 |
0.467 |
0.453 |
0.021 |
0.035 |
 |
| | Fig. 12 The HOMO–LUMO gap of: receptors DPE-I and DPE-II and their Fe3+ complexes calculated at the B3LYP/6-31G/LANL2DZ level respectively. | |
Binding energies obtained from theoretical studies (as shown in Table S2†) have proved that energy values of DPE molecules decrease with the addition of Fe3+. Energy for DPE-I decreases from −1175 a.u to −1298 a.u while for DPE-II energy decrease found was from −1177 a.u to −1299 a.u. Hence, leading to the formation of the stable complexes. Energy decrease in case of DPE-II is slightly more than that of DPE-I proving DPE-II to be more stable than DPE-I.
Real life sampling
In order to test the analytical applicability of the proposed voltammetric method, the chemosensors were used for the determination of iron in pharmaceutical samples (Fer-iron) and water samples (tap water and mineral water) and some synthetic samples.
Pharmaceutical sample was prepared by dissolving one tablet of Fer-iron in 10 mL HCl and heated to dryness. After that, the sample was dissolved in 10 mL distilled water, filtered and transferred to a 25 mL standard flask and this volume was completed with distilled water. Each synthetic sample (RS-1 and RS-2) was spiked with the addition of 1 mL of 10−2 M standard metal ion solution, keeping the total volume of the sample equal to 100 mL. Complexometric method was also used for the determination of iron contents in these samples. The results of the voltammetric sensors obtained are presented in Table 7 and compared with those obtained by using complexation with EDTA. The sensors are found to be in satisfactory agreement with those obtained from the complexometric method. These observations and results have confirmed that present chemosensors can be used for practical analysis.
Table 7 Determination of Fe(III) in real samples using complexometric method and proposed chemosensorsa
| Sample name |
Adjusted pH |
Total iron found (μg mL−1 or g kg−1) |
| Chemosensor (DPE-1) |
Complexometric method |
Chemosensor (DPE-II) |
Complexometric method |
| ± shows the standard deviation (n = 3). |
| Fer-iron |
3.5 |
10.7 ± 0.02 |
11.1 ± 0.03 |
12.5 ± 0.02 |
12.2 ± 0.01 |
| Tap water |
3.5 |
6.1 ± 0.08 |
6.4 ± 0.06 |
6.59 ± 0.07 |
7.1 ± 0.02 |
| Mineral water |
3.5 |
5.3 ± 0.01 |
5.1 ± 0.01 |
4.89 ± 0.06 |
4.9 ± 0.02 |
| RS-1 |
3.5 |
11.53 ± 0.03 |
11.4 ± 0.03 |
12.79 ± 0.05 |
12.7 ± 0.01 |
| RS-2 |
3.5 |
15.39 ± 0.02 |
15.5 ± 0.04 |
16.89 ± 0.05 |
16.9 ± 0.04 |
Acknowledgements
We thank BRNS, BARC Mumbai for financial assistance vide research grant no. 2012/37C/5/BRNS/621 and we also thank SAI Labs, Thapar University, Patiala for NMR studies.
References
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS PubMed.
- C. Yi, W. Tian, B. Song, Y. Zheng, Z. Qi, Q. Qi and Y. Sun, J. Lumin., 2013, 141, 15–22 CrossRef CAS PubMed.
- L. Fabbrizzi and A. Poggi, Chem. Soc. Rev., 1995, 24, 197–202 RSC.
- M. R. Pinto and K. S. Schanze, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7505–7510 CrossRef CAS PubMed.
- I.-B. Kim, A. Dunkhorst, J. Gilbert and U. H. Bunz, Macromolecules, 2005, 38, 4560–4562 CrossRef CAS.
- N. Singh, N. Kaur, J. Dunn, M. MacKay and J. F. Callan, Tetrahedron Lett., 2009, 50, 953–956 CrossRef CAS PubMed.
- A. D. Child and J. R. Reynolds, Macromolecules, 1994, 27, 1975–1977 CrossRef CAS.
- M. R. Pinto, B. M. Kristal and K. S. Schanze, Langmuir, 2003, 19, 6523–6533 CrossRef CAS.
- K. K. Stokes, K. Heuzé and R. D. McCullough, Macromolecules, 2003, 36, 7114–7118 CrossRef CAS.
- S. Kamila, J. F. Callan, R. C. Mulrooney and M. Middleton, Tetrahedron Lett., 2007, 48, 7756–7760 CrossRef CAS PubMed.
- I.-B. Kim, J. N. Wilson and U. H. Bunz, Chem. Commun., 2005, 1273–1275 RSC.
- C. Xue, V. R. Donuru and H. Liu, Macromolecules, 2006, 39, 5747–5752 CrossRef CAS.
- P. B. Balanda, M. B. Ramey and J. R. Reynolds, Macromolecules, 1999, 32, 3970–3978 CrossRef CAS.
- B. S. Gaylord, A. J. Heeger and G. C. Bazan, J. Am. Chem. Soc., 2003, 125, 896–900 CrossRef CAS PubMed.
- H. Wu, F. Huang, Y. Mo, W. Yang, D. Wang, J. Peng and Y. Cao, Adv. Mater., 2004, 16, 1826–1830 CrossRef CAS.
- O. Fix and K. Kowdley, Minerva Med., 2008, 99, 605–617 CAS.
- P. Aisen, M. Wessling-Resnick and E. A. Leibold, Curr. Opin. Chem. Biol., 1999, 3, 200–206 CrossRef CAS.
- S. Altamura and M. U. Muckenthaler, J. Alzheimer's Dis., 2009, 16, 879–895 Search PubMed.
- L. Silvestri and C. Camaschella, J. Cell. Mol. Med., 2008, 12, 1548–1550 CrossRef CAS PubMed.
-
(a) A. L. Crumbliss and J. M. Harrington, Adv. Inorg. Chem., 2009, 61, 179 CrossRef CAS;
(b) M. S. Mitchell, D. L. Walker, J. Whelan and B. Bosnich, Inorg. Chem., 1987, 26, 396–400 CrossRef CAS.
- A. Ohashi, H. Ito, C. Kanai, H. Imura and K. Ohashi, Talanta, 2005, 65, 525–530 CrossRef CAS PubMed.
- I. Juranovic, P. Breinhoelder and I. Steffan, J. Anal. At. Spectrom., 2003, 18, 54–58 RSC.
- P. Ugo, L. Moretto, A. De Boni, P. Scopece and G. Mazzocchin, Anal. Chim. Acta, 2002, 474, 147–160 CrossRef CAS.
- E. Bakkaus, R. N. Collins, J.-L. Morel and B. Gouget, J. Chromatogr. A, 2006, 1129, 208–215 CrossRef CAS PubMed.
- C. M. van den Berg, Anal. Chem., 2006, 78, 156–163 CrossRef CAS PubMed.
- L. Tarazi, N. Narayanan, J. Sowell, G. Patonay and L. Strekowski, Spectrochim. Acta, Part A, 2002, 58, 257–264 CrossRef.
- J. Mao, L. Wang, W. Dou, X. Tang, Y. Yan and W. Liu, Org. Lett., 2007, 9, 4567–4570 CrossRef CAS PubMed.
- S. Ghosh, R. Chakrabarty and P. S. Mukherjee, Inorg. Chem., 2008, 48, 549–556 CrossRef PubMed.
- A. J. Weerasinghe, C. Schmiesing, S. Varaganti, G. Ramakrishna and E. Sinn, J. Phys. Chem. B, 2010, 114, 9413–9419 CrossRef CAS PubMed.
- L.-F. Zhang, J.-L. Zhao, X. Zeng, L. Mu, X.-K. Jiang, M. Deng, J.-X. Zhang and G. Wei, Sens. Actuators, B, 2011, 160, 662–669 CrossRef CAS PubMed.
- B. Ma, S. Wu and F. Zeng, Sens. Actuators, B, 2010, 145, 451–456 CrossRef CAS PubMed.
- S.-R. Liu and S.-P. Wu, Sens. Actuators, B, 2012, 171, 1110–1116 CrossRef PubMed.
- Q. Meng, W. Su, C. He and C. Duan, Talanta, 2012, 97, 456–461 CrossRef CAS PubMed.
- V. Bravo, S. Gil, A. M. Costero, M. N. Kneeteman, U. Llaosa, P. M. Mancini, L. E. Ochando and M. Parra, Tetrahedron, 2012, 68, 4882–4887 CrossRef CAS PubMed.
- Y. R. Bhorge, H.-T. Tsai, K.-F. Huang, A. J. Pape, S. N. Janaki and Y.-P. Yen, Spectrochim. Acta, Part A, 2014, 130, 7–12 CrossRef CAS PubMed.
- C. Queirós, A. M. Silva, S. C. Lopes, G. Ivanova, P. Gameiro and M. Rangel, Dyes Pigm., 2012, 93, 1447–1455 CrossRef PubMed.
- D. Staneva, P. Bosch and I. Grabchev, J. Mol. Struct., 2012, 1015, 1–5 CrossRef CAS PubMed.
- N. R. Chereddy, M. V. N. Raju, P. Nagaraju, V. R. Krishnaswamy, P. S. Korrapati, P. R. Bangal and V. J. Rao, Analyst, 2014, 139, 6352–6356 RSC.
- D. Shi, M. Ni, J. Luo, M. Akashi, X. Liu and M. Chen, Analyst, 2015, 140, 1306–1313 RSC.
- H. Xu, S. Zhou, L. Xiao, H. Wang, S. Li and Q. Yuan, J. Mater. Chem. C, 2015, 3, 291–297 RSC.
- R. R. Koner, S. Sinha, S. Kumar, C. K. Nandi and S. Ghosh, Tetrahedron Lett., 2012, 53, 2302–2307 CrossRef CAS PubMed.
- A. Kamal, N. Kumar, V. Bhalla, M. Kumar and R. K. Mahajan, Sens. Actuators, B, 2014, 190, 127–133 CrossRef CAS PubMed.
- Z.-Q. Liang, C.-X. Wang, J.-X. Yang, H.-W. Gao, Y.-P. Tian, X.-T. Tao and M.-H. Jiang, New J. Chem., 2007, 31, 906–910 RSC.
- M. Chhibber, G. Kumar, P. Parasuraman, T. Ramya, N. Surolia and A. Surolia, Bioorg. Med. Chem., 2006, 14, 8086–8098 CrossRef CAS PubMed.
- P. Goswami and D. K. Das, J. Fluoresc., 2012, 22, 1081–1085 CrossRef CAS PubMed.
- J.-Q. Wang, L. Huang, M. Xue, Y. Wang, L. Gao, J. H. Zhu and Z. Zou, J. Phys. Chem. C, 2008, 112, 5014–5022 CAS.
- A. Senthilvelan, I. Ho, K. C. Chang, G. H. Lee, Y. H. Liu and W. S. Chung, Chem.–Eur. J., 2009, 15, 6152–6160 CrossRef CAS PubMed.
- D. Schaming, C. Costa-Coquelard, I. Lampre, S. Sorgues, M. Erard, X. Liu, J. Liu, L. Sun, J. Canny and R. Thouvenot, Inorg. Chim. Acta, 2010, 363, 2185–2192 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS PubMed.
- F. Wang, R. Nandhakumar, J. H. Moon, K. M. Kim, J. Y. Lee and J. Yoon, Inorg. Chem., 2011, 50, 2240–2245 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra00969c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.