
Synthesis and biological evaluation of non-glucose glycoconjugated N-hydroyxindole class LDH inhibitors as anticancer agents†
Valeria Di Bussoloa,
Emilia C. Calvaresi‡
b,
Carlotta Granchia,
Linda Del Binoa,
Ileana Fraua,
Maria Chiara Dasso Lang§
a,
Tiziano Tuccinardia,
Marco Macchiaa,
Adriano Martinellia,
Paul J. Hergenrother*b and
Filippo Minutolo*a
aDipartimento di Farmacia, Università di Pisa, Via Bonanno 33, 56126 Pisa, Italy. E-mail: filippo.minutolo@farm.unipi.it
bDepartment of Chemistry, University of Illinois, 600 S. Mathews Avenue, Urbana, IL 61801, USA. E-mail: hergenro@illinois.edu
First published on 11th February 2015
Abstract
Inhibitors of human lactate dehydrogenase A (LDH-A) are promising therapeutic agents against cancer. The development of LDH-A inhibitors that possess cellular activities has so far proved to be particularly challenging, since the enzyme's active site is narrow and highly polar. In the recent past, we were able to develop a glucose-conjugated N-hydroxyindole-based LDH-A inhibitor designed to exploit the sugar avidity expressed by cancer cells (the Warburg effect). Herein we describe a structural modulation of the sugar moiety of this class of inhibitors, with the insertion of α-D-mannose, β-D-gulose, or β-N-acetyl-D-glucosamine portions in their structures. Their stereospecific chemical synthesis, which involves a substrate-dependent stereospecific glycosylation step, and their biological activity in reducing lactate production and proliferation in cancer cells are reported. Interestingly, the α-D-mannose conjugate displayed the best properties in the cellular assays, demonstrating an efficient antiglycolytic and antiproliferative activity in cancer cells.
Introduction
Tumors generally follow unusual metabolic pathways to obtain the energy and anabolites required for their persistent growth. Growing cancer cells rewire their metabolism to meet their bioenergetic and biosynthetic needs for rapid proliferation by increasing their consumption of glucose and glutamine, enhancing glycolysis, and increasing the excretion of lactate. In particular, glycolysis, rather than oxidative phosphorylation (OXPHOS), is predominantly used even in the presence of oxygen, as described by the well-known “Warburg effect”.1,2 In fact, in normal cells, most of the pyruvate produced by glycolysis goes into the tricarboxylic acid (TCA) cycle and is eventually oxidized to CO2 via OXPHOS. On the contrary, tumor cells mostly convert pyruvate to lactate anaerobically. Several prospective drugs have been developed to take advantage of this peculiar metabolism occurring in cancer by acting as anti-glycolytic agents, as reviewed recently.3,4Lactate dehydrogenase (LDH), the enzyme that catalyzes the interconversion of pyruvate and lactate, establishes a key checkpoint for the switch from aerobic to anaerobic glycolysis. LDH is a tetrameric enzyme that may exist in five isoforms (hLDH1-5), which results from the possible combinations of the two subunits: LDH-A and LDH-B. Subunit LDH-A (and, consequently, its tetrameric functional form hLDH5 or LDH-A4) is very frequently found to be overexpressed in invasive cancer; its genetic silencing has been shown to reduce proliferation and invasiveness of tumor cells, especially under hypoxic conditions.5 The validity of LDH-A as an anticancer target is further strengthened by the fact that individuals homozygous for LDH-A deficiency do not show any particular clinical symptoms, except for myoglobinuria upon intense physical exertion.6 Therefore, several recent research efforts have been dedicated to the development of new LDH-A inhibitors with therapeutic potential as anticancer drugs, although problems related to poor cellular activities of these inhibitors have been often reported.7,8
The high rate of glycolysis in cancer cells is associated with a striking glucose avidity. This feature has been exploited by glycoconjugation of anticancer drugs, in order to improve their targeting to tumor cells versus normal tissues.9 We have previously developed a novel class of N-hydroxyindole (NHI)-based LDH-A inhibitors,10,11 and we recently reported that gluco-conjugated NHIs, in particular NHI-Glc-2 (1, Fig. 1), showed a remarkably improved potency in reducing the cellular production of lactic acid in cancer cells.12 Anticancer glycoconjugates are not limited to D-glucose; other sugar and aminosugar moieties have been previously introduced in anticancer agents with improved selectivity. This is due to the fact that glucose transporters overexpressed by tumor tissues, such as GLUT1, are able to transport a number of sugar substrates besides glucose.9 Therefore, the ability of glucose analogues, such as α-manno-2 and β-gulo-conjugate 3, to be efficiently taken up by cancer cells, results in an attractive strategy of extending our “dual targeting” of the Warburg effect.12 to a wider class of sugar conjugates. Compound 2 is a conjugate of D-mannose, a C2 epimer of D-glucose; D-mannose is found at concentrations of around 50 μM in human serum, and it is primarily used in the glycosylation of proteins (specifically, mannose-6-phosphate is a localization tag applied to proteins destined for lysosomal import). Mannose is known to be transported into mammalian cells primarily by mannose-specific transporters normally expressed in intestinal cells; however, it has also been reported to be a good substrate for GLUT1 (ref. 13) with a 13-fold reduction of its affinity for this transporter (Km = 20 mM), when compared to that of glucose (Km = 1.5 mM).14 Compound 3 contains D-gulose, which is a stereoisomer of glucose differing in stereochemistry at the C3 and C4 positions; it is used by some archaea but it is not known to be found in human serum or used in human metabolism. So far, the ability of D-gulose to be transported by GLUTs has not been reported. In addition, since cancer cells have been found to utilize O-GlcN-acylation as a post-translational modification,15,16 we also synthesized compound 4, in which the NHI pharmacophoric unit is conjugated with GlcNAc.
 | ||
| Fig. 1 Chemical structures of the NHI-glycoconjugates: β-D-Glc-(1), α-D-Man-(2), β-D-Gul-(3), and β-D-Glc(NAc)-(4) NHIs. | ||
Herein, we describe the application of our originally-developed glycosylation protocols to the synthesis of glycoconjugates 2–4 (Fig. 1) and their biological evaluations as prospective anticancer agents interfering with the cellular production of lactic acid.
Results and discussion
Synthesis of the glycoconjugates
Recently, we reported a new glycosylation process, using glycal derived vinyl epoxides 6α and 6β as new glycosyl donors (Scheme 1). These systems, in the presence of alcohols or alkyl lithium reagents as nucleophiles, afford only 6-O-benzyl-protected O- and C-glycosides with the same configuration of the starting epoxide, in an uncatalyzed substrate-dependent stereospecific process.17–19 The driving force of this regio- and stereoselective 1,4-addition process is the occurrence of a coordination between the nucleophile and the oxirane oxygen in the form of hydrogen bond, in the case of O-nucleophile, or through the metal lithium cation, in the case of alkyl lithium reagents. Vinyl epoxides 6α and 6β cannot be isolated, because they are unstable and, therefore, they must be prepared only in situ by cyclization under basic conditions with t-BuOK of their ultimate precursor, the corresponding trans-hydroxymesylates 5 and 7.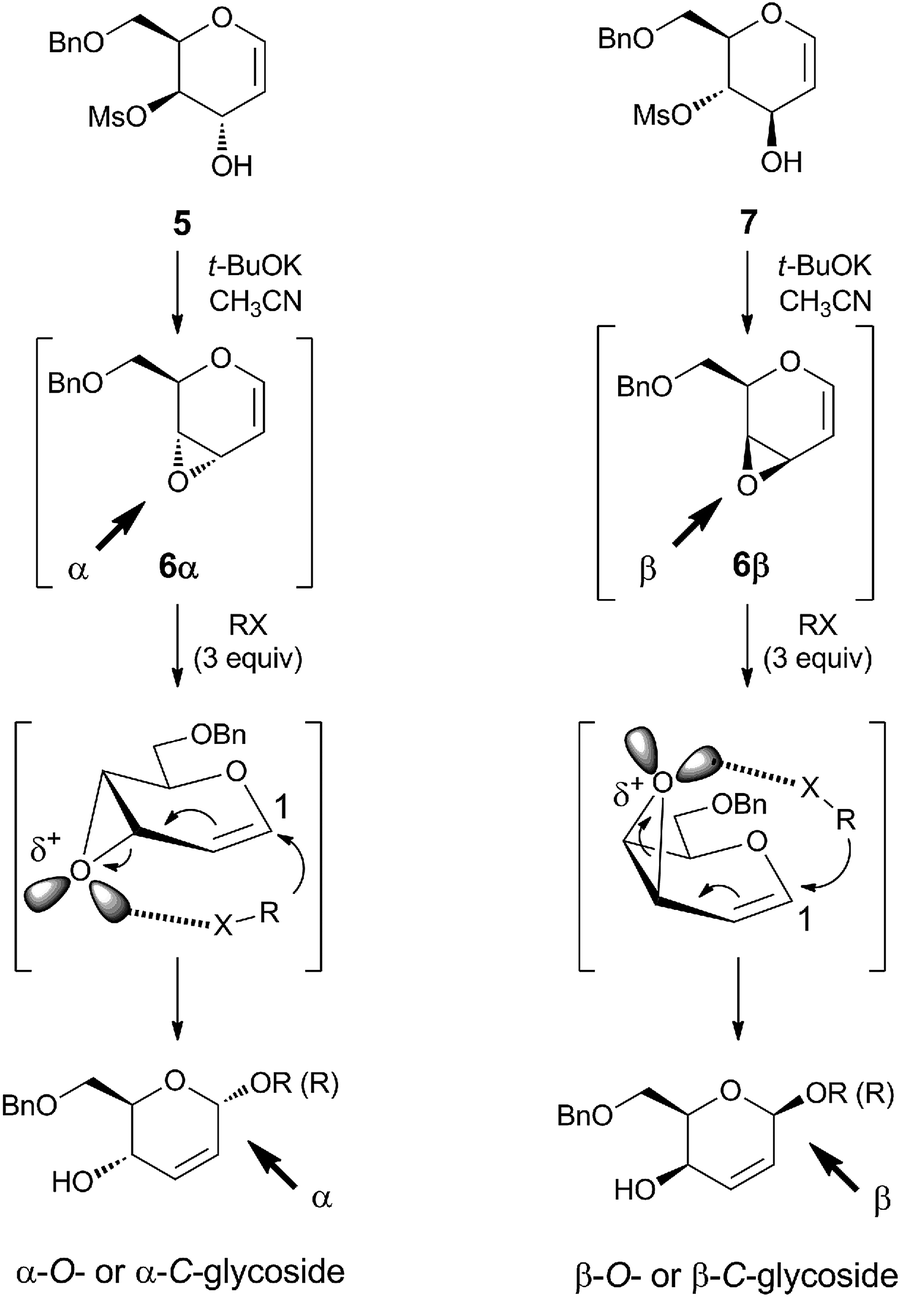 | ||
| Scheme 1 Substrate-dependent stereospecific glycosylation: reaction conditions and mechanism of the stereochemical outcome [O-nucleophiles (alcohols), X = OH; C-nucleophiles (lithium alkyls), X = Li; R = Me, Et, i-Pr, t-Bu, Ph, monosaccharides]. | ||
In the synthesis of glycoconjugates 2 and 3, due to the instability the N–O single bond under the reductive conditions utilized for the final removal of the benzyl protective group which was present in our original methodology (see 6α and 6β, Scheme 1), we decided to use synthetic intermediates in which the primary hydroxy functionality is protected as a tetrahydropyranyl ether (–OTHP), a protective group which can be easily removed by acid hydrolysis. The key intermediates are represented by THP-protected hydroxymesylate 8, which was synthesized in six steps starting from tri-O-acetyl-D-glucal as previously reported20 and 9, which was instead specifically synthesized from 8 (Scheme 2). The conversion of 8 to 9 started with a cyclization with t-BuOK in CH3CN, leading to the in situ formation of epoxide 10β, which was followed by a ring opening reaction with a non-coordinating hydroxy ion equivalent, such as Me3SiO− present in Bu4N+Me3SiO−, a salt prepared by addition of potassium trimethyl silanolate (Me3SiOK) to a solution of tetrabutyl ammonium bromide (TBAB) in THF.19 In this way, after exposure to water, a clean 1,2-addition process is obtained, affording the desired trans diol 12, which was treated with TBSCl to give the mono allyl C(3)-O-TBS derivative 13. Subsequent mesylation of 13 at C(4) produced the all-protected glycal derivative 14, which was finally deprotected with TBAF/THF to give trans hydroxymesylate 9.
 | ||
| Scheme 2 Synthesis of THP-protected trans-hydroxymesylates 8 and 9 from tri-O-acetyl-D-glucal. | ||
The synthesis of β-gulo-derivative 3 started from trans-hydroxymesylate 8 (Scheme 3), with the in situ formation of vinyl epoxide 10β by cyclization with t-BuOK in CH3CN. This reactive intermediate was not isolated. It was immediately treated with the NHI-based glycosyl acceptor 15 (NHI-2, a LDH-A inhibitor previously reported by us11) to give, after only 30 minutes at room temperature, the glycosylation product 16 with complete 1,4-regio- and β-stereoselectivity. Functionalization of the double bond present in 16 by OsO4/NMO protocol afforded the corresponding syn-dihydroxylated product 18, in accordance with a complete sterically-favored α-facial stereoselective electrophilic addition (70% yield). The deprotection of the C(6)-OTHP functionality of 18 by using catalytic PPTS in absolute EtOH under controlled temperature at 40 °C for 46 h, afforded the desired β-O-D-gulo-conjugate 3 in good yield after recrystallization (75% yield). A similar protocol was utilized for the preparation of the α-manno-derivative 2 (Scheme 3). In this case, the reaction sequence started from trans-hydroxymesylate 9 with a cyclization reaction with t-BuOK in CH3CN to produce highly reactive vinyl epoxide 10α, which was treated in situ with glycosyl acceptor 15. Here again, the glycosylation step proved to be very efficient after only 30 minutes at room temperature, with the production of α-glycoconjugate 17 with a complete 1,4-regio- and α-stereoselectivity (58% yield after purification). Once obtained, the unsaturated α-glycoconjugate 17 was submitted to cis-dihydroxylation with OsO4/NMO and, in this case, the β-stereoselective attack of the electrophile is directed by the allyl substituents at C(4) and C(1), now located on the α-face. Therefore the corresponding β-dihydroxylated derivative 19 was the only stereoisomer obtained (65% yield). The final deprotection of the THP moiety present at C(6) of 19, carried out in absolute EtOH for 20 h at 40 °C in the presence of PPTS, provided the α-O-D-manno-conjugate 2 in 80% yield after recrystallization.
 | ||
| Scheme 3 Synthesis of glycoconjugates 2 and 3 by stereospecific glycosylation of NHI-derivative 15. | ||
The synthesis of GlcNAc-conjugate 4 (Scheme 4) utilized as the glycosyl donor oxazoline 20, which was prepared as previously reported.21 Glycosyl acceptor 15 displayed a low reactivity as the nucleophile with oxazoline 20 and, therefore, we needed to test a wide array of reaction conditions. The best results were obtained by using TMSOTf as the catalyst.22 This way, the ring opening of oxazoline 20 was realized in anhydrous dichloroethane at 80 °C for 24 h in the presence of molecular sieves 4 Å using 0.5 equiv of TMSOTf and 1.5 equiv of NHI 15 to afford peracetylated Glc-NAc-conjugate 21 in 58% yield after purification. Deacetylation of 21 with MeONa in CH2Cl2/MeOH afforded Glc-NAc-conjugate 4 in quantitative yields.
 | ||
| Scheme 4 Synthesis of GlcNAc conjugate 4 via the formation of oxazoline 20 and subsequent stereospecific glycosylation of NHI-derivative 15. | ||
Enzyme inhibition assays
The inhibitory activities of the sugar conjugates 2–4 were measured by standard enzyme kinetic experiments on purified human enzyme isoform hLDH5. The Ki values of each compound (Table 1) were measured in NADH-competition experiments as previously described.12| Compound | LDH-A – Ki (μM) |
|---|---|
| a Ki values were obtained by non-linear regression analysis with GraphPad Prism software (GraphPad, La Jolla, CA) using a second order polynomial regression analysis by applying the mixed-model inhibition fit (mean values ± SD calculated from at least 2 experiments, see Experimental section).b From ref. 12. | |
| 1 | 37.8 ± 0.9b |
| 2 | 92.4 ± 9.0 |
| 3 | 68.1 ± 17.5 |
| 4 | 98.4 ± 15.6 |
All the newly synthesized sugar conjugates (2–4) proved to be moderately active in these assays, with Ki values ranging from 68.1 μM with the manno-derivative 3 to 98.4 μM with the amino–glucose-derivative 4. None of them proved to be more potent than glucose-conjugate 1 (ref. 12) in these in vitro assays on the isolated enzyme.
Molecular modeling
Docking studies followed by molecular dynamic (MD) simulations were carried out to examine the putative modes of interaction of sugar conjugates 2–4 with LDH-A and the results of these studies are shown in Fig. 2. | ||
| Fig. 2 MD simulation results for the complex of LDH-A with 2 (A), 3 (B), and 4 (C). | ||
As highlighted in Fig. 2, all three compounds show a very similar positioning into the LDH-A binding site. In all cases, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O portion of the ester group of these compounds forms an H-bond with R169, and the methyl group of the ester establishes lipophilic interactions with the isopropyl side chain of V235. The 4-(trifluoromethyl)indole central scaffold is placed in a cleft mainly delimited by H193, G194, A238, V241, and I242, whereas the 6-phenyl group is directed toward the entrance of the binding site cavity. The sugar moiety is always placed in the NADH-binding pocket but, depending on the type of monosaccharide attached, it shows different H-bonds with the protein. Specifically, the α-mannose ring of 2 (Fig. 2A) forms two H-bonds with the hydroxyl group of T248 through its 2- and 3-hydroxyl groups. The β-gulose ring of 3 (Fig. 2B) shows a slightly different interaction mode with the protein, with a H-bond occurring between its 4-OH group and T248, and a peculiar H-bond that is formed by its 6-OH group and the side chain of N138. Finally, the GlcNAc portion of 4 (Fig. 2C) forms completely different interactions with the enzyme active site, the most important of which seem to be the interaction of the acetamido-group with T248, through its N–H portion, and with R169 through its CO moiety.
O portion of the ester group of these compounds forms an H-bond with R169, and the methyl group of the ester establishes lipophilic interactions with the isopropyl side chain of V235. The 4-(trifluoromethyl)indole central scaffold is placed in a cleft mainly delimited by H193, G194, A238, V241, and I242, whereas the 6-phenyl group is directed toward the entrance of the binding site cavity. The sugar moiety is always placed in the NADH-binding pocket but, depending on the type of monosaccharide attached, it shows different H-bonds with the protein. Specifically, the α-mannose ring of 2 (Fig. 2A) forms two H-bonds with the hydroxyl group of T248 through its 2- and 3-hydroxyl groups. The β-gulose ring of 3 (Fig. 2B) shows a slightly different interaction mode with the protein, with a H-bond occurring between its 4-OH group and T248, and a peculiar H-bond that is formed by its 6-OH group and the side chain of N138. Finally, the GlcNAc portion of 4 (Fig. 2C) forms completely different interactions with the enzyme active site, the most important of which seem to be the interaction of the acetamido-group with T248, through its N–H portion, and with R169 through its CO moiety.
Cellular lactate production inhibition assays
The ability of the glycol-conjugated NHIs 2–4 to inhibit lactate production in cells was evaluated using the same previously reported technique developed for glucose-conjugate 1,12 and the results are displayed in Fig. 3. | ||
| Fig. 3 Lactate production inhibition of glycoconjugates 1–4. HeLa human cervical carcinoma cells were treated under normoxic conditions with indicated compound concentrations or 1% DMSO vehicle, in DMEM supplemented with 10 mM unlabeled glucose, 1 mM pyruvate, and 4 mM glutamine. Following 8 h incubation, medium aliquots were collected, concentrated, derivatized using MTBSTFA + 1% TBDMCS catalyst, and assessed by GC-MS. Lactate was normalized in each sample using the 1 mM chlorophenylalanine (CPA) internal standard, and lactate relative to vehicle is depicted. Averages from three or more independent experiments are depicted, with error bars depicting standard error (n ≥ 3). | ||
In testing these glyco-conjugates at 50–200 μM concentrations for their ability to reduce lactate production in HeLa cells following an 8 h incubation, it was found that the α-manno-derivative 2 led to a potent, dose-dependent reduction in lactate production, which is comparable to that obtained with glucose-conjugate 1, with a remarkable 55% inhibition at the lowest concentration used (50 μM). A noticeable dose-dependent activity was also found with β-gulo derivative 3, although it was lower than those observed with 1 and 2. On the contrary, GlcNAc-conjugate 4 lacked any significant activity in this assay, and, therefore, was not considered in our subsequent experiments. Under these conditions, a very modest effect is observed with very high concentrations (10 mM) of hexokinase inhibitor 2-deoxyglucose, whereas negligible effects are observed upon incubation with 10 μM of the topoisomerase II inhibitor etoposide, a cytotoxic compound that does not affect glucose metabolism.
Cellular uptake assays
The relative cell uptake levels of glyco-conjugates 2 and 3, and of reference glucose-conjugate 1,12 versus their aglycone counterpart NHI-2 (15, Scheme 3), and the potential cleavage of the mannose-(2) and gulo-portions (3) in cell culture, was assessed. A549 cells were treated with 100 μM concentrations of each compound for 4 h. After washing and lysing the cells, the cellular fraction of each compound was assessed by LC-MS and correlated to concentration using calibration standards (Fig. S1, ESI†), and the intracellular concentrations are displayed in Fig. 4. | ||
| Fig. 4 Results of the intracellular concentration comparison study comparing relative uptake of NHI-2 (15) to its three glycoconjugates 1–3. A549 non-small cell lung carcinoma cells were treated with 100 μM concentrations of each compound for 4 h; cells were then lysed, sonicated in methanol, and assessed for intracellular compound concentration by LC-MS. Triplicate data is shown, with error bars denoting standard error. Statistical analysis was performed using an unpaired Student's t test to compare glycoconjugate intracellular concentrations with NHI-2 intracellular concentration, with * denoting p < 0.05, ** denoting p < 0.01, and *** denoting p < 0.0005. | ||
As we had previously observed with glucose-conjugate 1,12 mannose-derivative 2 displays a significantly enhanced cell uptake compared to the aglycone NHI-2. While 1 and 2 had similar levels of cell uptake, the gulose-derivative 3 had slightly lower levels of cell uptake. These results are consistent with those obtained in the cellular lactate production inhibition assays (Fig. 3), where the most potent cellular inhibitors were found to be 1 and 2, thus positively correlating cellular activity and cell uptake. It is important to note that no cleavage of any of these compounds was observed in either the UV trace or TIC of the resultant lysate samples from 4 h of incubation in A549 cells, so all the effects should be ascribed to the parent compounds.
Cancer cell antiproliferative potency assays
After assessing the ability of glycol-conjugates 2 and 3 to penetrate cells and inhibit cellular lactate production, their potency in killing HeLa and A549 cancer cells was evaluated and compared to that of their glucose analogue 1 and of their aglycone NHI-2 (15). The growth inhibitory effect of these compounds was evaluated by treating the cancer cells for 72 h with varying concentrations of the compounds, after which cell death was assessed by the Sulforhodamine B (SRB) assay.23 The antiproliferative potencies are expressed as IC50 values (the concentration of compound required to kill 50% of cells, where lower concentrations indicate increased potency) and are presented in Table 2.| Compound | HeLa (cervical carcinoma) | A549 (NSCLC) |
|---|---|---|
| a Three independent experiments were performed under normoxic conditions. Remaining biomass after fixing with 10% trichloroacetic acid was quantified by sulforhodamine B staining.b Data from ref. 11.c Data from ref. 12. | ||
| NHI-2 (15) | 33.4 ± 1.0b | 44.1 ± 6.2b |
| 1 | 7.2 ± 0.2c | 17.2 ± 3.0c |
| 2 | 5.4 ± 1.3 | 15.2 ± 0.7 |
| 3 | 11.8 ± 0.1 | 24.7 ± 0.9 |
Glycoconjugated NHI compounds 1–3 were all more potent in killing cancer cells than their respective aglycone (NHI-2, 15), in agreement with their increased cell uptake (Fig. 4) and increased cellular lactate production inhibition (Fig. 3) compared to the aglycone. In particular, the newly-synthesized compounds have 3-6-fold (compound 2) and 2-3-fold (compound 3) enhanced potencies compared to NHI-2. Finally, the highest potency levels were found with the new mannose-derivative 2, which is even more potent than glucose-derivative 1, and shows IC50 values of 5.4 μM against HeLa cells and 15.2 μM against A549 cells.
Conclusions
We have successfully planned and implemented the stereospecific synthesis of three new glycoconjugates (2–4), which were intended to extend the chemical class of sugar-conjugates of NHIs. These glycoconjugates were designed to be LDH-A inhibitors with efficient cell uptake. The biological assays in cancer cells displayed a remarkable correlation between the cell uptake, reduction of cellular production of lactate, and inhibition cellular proliferation. Overall, the α-manno-derivative 2 proved to be the most efficient compound in cell-based assays. These results confirm the validity of the dual targeting strategy of the Warburg effect and pave the way to a more expansive exploration of glycoconjugates, both as molecular tools and as potential therapeutic agents that block tumor glycolysis.Experimental
Materials and methods
All solvents and chemicals were used as purchased without further purification. Chromatographic separations were performed on silica gel columns by flash (Kieselgel 40, 0.040–0.063 mm; Merck) or gravity column (Kieselgel 60, 0.063–0.200 mm; Merck) chromatography. Reactions were followed by thin-layer chromatography (TLC) on Merck aluminum silica gel (60 F254) sheets that were visualized under a UV lamp. Evaporation was performed in vacuo (rotating evaporator). Sodium sulfate was always used as the drying agent. Proton (1H) and carbon (13C) NMR spectra were obtained with a Bruker Avance III 400 MHz or Bruker Avance 250 MHz spectrometer using the indicated deuterated solvents. Chemical shifts are given in parts per million (ppm) (δ relative to residual solvent peak for 1H and 13C). Yields refer to isolated and purified products. LC-MS characterization and high-resolution mass spectrometry (HRMS) analysis were performed using a Waters Quattro II quadrupole–hexapole–quadrupole liquid chromatography/mass spectrometry apparatus (Waters, Milford, MA) equipped with an electrospray ionization source. LC separation was achieved using a C18 Waters Xbridge column (2.1 × 20 mm, Waters) at 25 °C using a linear gradient of mobile phases: 95% H2O, 5% acetonitrile, and 0.1% formic acid (A) and 95% acetonitrile, 5% H2O, and 0.1% formic acid (B). Solution A was initially passed through the column but decreased linearly to 50% of the mobile phase at 10 minutes and 0% of the mobile phase at 25 minutes. The flow rate was 200 μL min−1, and the injection volume was 10 μL. The ultraviolet (UV) detector was programmed to monitor absorbance at 254 nm, to detect the phenyl ring present in all compounds.Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexane–AcOEt (0.1% Et3N) mixture afforded pure glycoside 16 (0.206 g, 77% yield) as a liquid. Rf = 0.33 (4:6 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 8.13–8.27 (m corresponding to two diastereoisomers, overall 1H; Ar), 7.62–7.96 (m, 3H; Ar), 7.15–7.52 (m, 4H; Ar), 6.23–6.47 (m corresponding to two diastereoisomers, overall 2H; vinyl CH), 5.96–6.05 (m corresponding to two diastereoisomers, overall 1H; anomeric CH), 4.31–4.59 (m corresponding to two diastereoisomers, overall 1H; THP–CHO2), 3.08–4.15 (m corresponding to two diastereoisomers, overall 6H; CHO & CH2O), 3.96 (s, 3H; COOCH3), 1.29–1.74 (m corresponding to two diastereoisomers, overall 6H; THP–CH2).:1 t-BuOH–acetone mixture (0.31 mL) was added to a 50% p/v aqueous solution of N-methyl morpholine-N-oxide (NMO) (80 μL) and the resulting reaction mixture was treated with 2.5% p/v OsO4 solution in t-BuOH (80 μL) and stirred for 2 h at the same temperature. Dilution with AcOEt and evaporation of the filtered (Celite®) organic solution afforded a crude reaction product (0.092 g), which was subjected to flash chromatography. Elution with a 2:8 hexane–AcOEt (0.1% Et3N) mixture afforded 18 (0.037 g, 70% yield), practically pure as a liquid. Rf = 0.12 (2:8 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 8.25–8.31 (m corresponding to two diastereoisomers, overall 1H; Ar), 7.58–7.79 (m, 3H; Ar), 7.34–7.54 (m, 4H; Ar), 5.37–5.49 (m corresponding to two diastereoisomers, overall 1H; anomeric CHO), 5.14–5.25 (m corresponding to two diastereoisomers, overall 1H; THP–CHO2), 3.90–4.46 (m corresponding to two diastereoisomers, overall 5H; CH2O and CHO), 3.98 (s, 3H; COOMe), 3.34–3.80 (m corresponding to two diastereoisomers, overall 3H; CH2O and CHO), 1.40–1.74 (m corresponding to two diastereoisomers, overall 6H; THP–CH2).:1 hexane–acetone). [α]20D: +57.71 (c 0.35, CH3OH). 1H NMR (400 MHz; CD3OD) δ 8.31–8.36 (m, 1H; Ar), 7.72–7.79 (m, 3H; Ar), 7.45–7.54 (m, 2H; Ar), 7.40 (tt, 1H, J = 7.4, 1.2 Hz; Ar), 7.23 (qd, 1H, J = 1.8, 0.9 Hz; Ar), 5.47 (d, 1H, J = 8.3 Hz; anomeric CHO), 4.11 (t, 1H, J = 3.4 Hz; CHO), 4.03 (dd, 1H, J = 8.3, 3.4 Hz; CHO), 3.98 (s, 3H; COOMe), 3.93 (td, 1H, J = 6.2, 1.1 Hz; CHO), 3.81–3.85 (m, 1H; CHO), 3.79 (dd, 1H, J = 10.8, 4.6 Hz; CH2O), 3.66 (dd, 1H, J = 10.8, 6.1 Hz; CH2O). 13C NMR (62.5 MHz; CD3OD) δ 162.2, 141.2, 140.1, 139.9, 130.1 (2C), 129.8, 129.1, 128.4 (2C), 125.9 (q, J = 271.7 Hz), 124.4 (q, J = 32.3 Hz), 120.1 (q, J = 4.6 Hz), 118.4 (q, J = 1.3 Hz), 115.2, 108.8, 106.7, 75.5, 73.2, 70.4, 68.6, 61.9, 52.4. HRMS: (M + H+) found 498.1370; C23H23F3NO8 requires 498.1376.:7 hexane–AcOEt (0.1% Et3N) mixture afforded pure trans diol 12 (0.235 g, 45% yield), as a colorless liquid. Rf = 0.13 (3:7 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 6.58–6.66 (m corresponding to two diastereoisomers, overall 1H, vinyl CHO), 4.95–5.05 (m corresponding to two diastereoisomers, overall 1H, vinyl CH), 4.64–4.72 (m corresponding to two diastereoisomers, overall 1H, THP–CHO2), 4.06–4.20 (m corresponding to two diastereoisomers, overall 1H; CHO), 3.77–4.04 (m corresponding to two diastereoisomers, overall 5H CHO and CH2O), 3.47–3.62 (m corresponding to two diastereoisomers, overall 1H; CHO), 1.69–1.85 (m corresponding to two diastereoisomers, overall 3H, THP–CH2), 1.48–1.68 (m corresponding to two diastereoisomers, overall 3H; THP–CH2).:2 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 6.49–6.58 (m corresponding to two diastereoisomers, overall 1H; vinyl CHO), 4.81–4.90 (m corresponding to two diastereoisomers, overall 1H; vinyl CH), 4.63–4.71 (m corresponding to two diastereoisomers, overall 1H; THP–CHO2), 3.95–4.17 (m corresponding to two diastereoisomers, overall 2H; CHO and CH2O), 3.72–3.94 (m corresponding to two diastereoisomers, overall 4H CHO and CH2O), 3.43–3.62 (m corresponding to two diastereoisomers, overall 1H, CHO), 1.44–1.86 (m corresponding to two diastereoisomers, overall 6H; THP–CH2), 0.87 (s, 9H; (CH3)3CSi), 0.10 (s, 6H; CH3Si).:2 hexane–AcOEt (0.1% Et3N) mixture afforded pure mesylate derivative 14 (0.254 g, 71% yield) as a yellow liquid. Rf = 0.19 (8:2 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 6.45–6.54 (m corresponding to two diastereoisomers, overall 1H; vinyl CHO), 4.82–4.91 (m corresponding to two diastereoisomers, overall 1H; vinyl CH), 4.58–4.76 (m corresponding to two diastereoisomers, overall 2H; CHOMs + THP–CHO2), 4.22–4.34 (m corresponding to two diastereoisomers, overall 1H; CHO), 4.15–4.21 (m corresponding to two diastereoisomers, overall 1H; CHO), 3.81–4.02 (m corresponding to two diastereoisomers, overall 2H; CH2O), 3.57–3.80 (m corresponding to two diastereoisomers, overall 1H; CHO), 3.43–3.56 (m corresponding to two diastereoisomers, overall 1H; CHO), 3.04–3.10 (m corresponding to two diastereoisomers, overall 3H; CH3SO2), 1.66–1.89 (m corresponding to two diastereoisomers, overall 2H; THP–CH2), 1.44–1.62 (m corresponding to two diastereoisomers, overall 4H; THP–CH2), 0.83–0.92 (m corresponding to two diastereoisomers, overall 9H; (CH3)3CSi), 0.09–0.16 (m corresponding to two diastereoisomers, overall 6H; CH3Si).:1 hexane–AcOEt) to produce pure trans-hydroxymesylate 9 (0.122 g, 67% yield) as a yellow liquid. Rf = 0.19 (1:1 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 6.53–6.61 (m corresponding to two diastereoisomers, overall 1H; vinyl CHO), 4.93–5.04 (m corresponding to two diastereoisomers, overall 1H; vinyl CH), 4.72–4.87 (m corresponding to two diastereoisomers, overall 1H; THP–CHO2), 4.59–4.68 (m corresponding to two diastereoisomers, overall 1H; CHOMs), 4.18–4.30 (m corresponding to two diastereoisomers, overall 2H; CH2O), 3.43–4.05 (m corresponding to two diastereoisomers, overall 4H CHO & CH2O), 3.04–3.13 (m corresponding to two diastereoisomers, overall 3H; CH3SO2), 1.36–1.90 (m corresponding to two diastereoisomers, overall 6H; THP–CH2).:1 hexane–AcOEt (0.1% Et3N) mixture afforded pure glycoside 17 (0.155 g, 58% yield) as a white solid. Rf = 0.33 (4:6 hexane–AcOEt); mp 50–53 °C; 1H NMR (250 MHz; CDCl3) δ 7.77–7.89 (m corresponding to two diastereoisomers, overall 1H; Ar), 7.59–7.76 (m corresponding to two diastereoisomers, overall 3H; Ar), 7.34–7.56 (m corresponding to two diastereoisomers, overall 3H; Ar), 7.27–7.33 (m corresponding to two diastereoisomers, overall 1H; Ar), 6.12–6.36 (m corresponding to two diastereoisomers, overall 2H, vinyl CH), 5.78–5.89 (m corresponding to two diastereoisomers, overall 1H; anomeric CHO), 3.26–4.53 (m corresponding to two diastereoisomers, overall 7H; CH2O and CHO), 3.94 (s, 3H; COOMe), 1.28–1.86 (m corresponding to two diastereoisomers, overall 6H; THP–CH2).:1 t-BuOH–acetone mixture (0.25 mL) was added to 50% p/v aqueous solution of N-methyl morpholine-N-oxide (NMO) (60 μL) and the resulting reaction mixture was treated with 2.5% p/v OsO4 solution in t-BuOH (60 μL) and stirred for 8 h at room temperature. Dilution with AcOEt and evaporation of the filtered (Celite®) organic solution afforded a crude reaction product which was purified by flash chromatography. Elution with a 2:8 hexane–AcOEt (0.1% Et3N) mixture afforded pure α-mannopyranoside 19 (0.028 g, 65% yield) as a white solid. Rf = 0.17 (2:8 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 7.76–7.88 (m corresponding to two diastereoisomers, overall 1H; Ar), 7.55–7.72 (m corresponding to two diastereoisomers, overall 3H; Ar), 7.32–7.50 (m corresponding to two diastereoisomers, overall 4H; Ar), 5.66–5.71 (m corresponding to two diastereoisomers, overall 1H; anomeric CHO), 4.66–4.77 (m corresponding to two diastereoisomers, overall 1H; THP–CHO2), 4.30–4.47 (m corresponding to two diastereoisomers, overall 2H; CHO), 3.93–4.22 (m corresponding to two diastereoisomers, overall 3H; CHO and CH2O), 3.90 (s, 3H; COOMe), 3.71–3.86 (m corresponding to two diastereoisomers, overall 2H; CH2O), 3.29–3.48 (m corresponding to two diastereoisomers, overall 1H; CHO), 1.30–1.71 (m corresponding to two diastereoisomers, overall 6H; THP–CH2).:1 AcOEt–acetone). [α]20D +18.14 (c 0.95, CH3OH). 1H NMR (400 MHz; CD3OD) δ 8.04 (s, 1H; Ar), 7.75–7.78 (m, 3H; Ar), 7.48–7.53 (m, 2H; Ar), 7.41 (tt, 1H, J = 7.4, 1.2 Hz; Ar), 7.25–7.29 (m, 1H; Ar), 5.57 (d, 1H, J = 1.9 Hz; anomeric CHO), 4.57–4.63 (m, 1H; CHO), 4.20–4.28 (m, 1H; CHO), 3.96 (s, 3H; COOMe), 3.83–3.89 (m, 4H; CH2O and CHO). 13C NMR (62.5 MHz; CD3OD) δ 161.3, 141.1, 140.5, 139.2, 130.2 (2C), 129.3, 129.1, 128.5 (2C), 125.2 (q, J = 271.0 Hz), 124.8 (q, J = 32.9 Hz), 120.2 (q, J = 4.9 Hz), 118.3 (q, J = 1.7 Hz), 113.6, 111.2, 106.9, 77.5, 72.4, 70.5, 67.9, 62.6, 52.8. HRMS: (M + H+) found 498.1366; C23H23F3NO8 requires 498.1376.:1 CHCl3/acetone mixture yielded pure compound 21 (0.140 g, yield 40%) as a pale yellow solid: mp 222–225 °C. [α]20D = +26.9 (c 0.79; CHCl3). Rf = 0.3 (9:1 CHCl3–acetone). 1H NMR (400 MHz; CDCl3): δ 8.10 (s, 1H; Ar), 7.74 (s, 1H; Ar), 7.63 (d, 2H, J = 7.7 Hz; Ar), 7.36–7.49 (m, 3H; Ar), 7.33 (bs, 1H; Ar), 6.70 (d, 1H, J = 8.6 Hz; NH), 5.52 (d, 1H, J = 9.05, anomeric CHO), 5.16–5.28 (m, 2H; CH2OAc), 4.55 (q, 1H, J = 9.0 Hz; CHOAc), 4.26 (dd, 1H, J = 12.4, 4.7 Hz; CHOAc), 3.98–4.08 (m, 1H; CHO), 3.95 (s, 3H; COOMe), 3.69–3.75 (m, 1H; CHN), 2.07 (s, 3H; CH3CO), 2.03 (s, 3H; CH3CO), 2.02 (s, 3H; CH3CO), 1.77 (s, 3H; CH3CO). 13C NMR (100 MHz; CDCl3): δ 171.0, 170.6 (2C), 169.4, 161.0, 140.2, 140.0, 139.6, 129.2 (2C), 128.3, 127.4 (2C), 124.3 (q, J = 272.4 Hz), 123.8 (q, J = 33.0 Hz), 120.0 (q, J = 4.8 Hz), 119.03 (q, J = 4.7 Hz), 117.4 (q, J = 2.1 Hz), 114.3, 108.4, 105.8, 73.6, 72.5, 68.0, 61.8, 52.5, 52.4, 23.3, 20.8, 20.7, 20.4.:3 mixture of CH2Cl2 and MeOH (4 mL) and cooled at 0 °C. A freshly prepared 0.33 M solution of MeONa/MeOH (0.05 mL) was added to the resulting solution and the reaction mixture was stirred for 4 h at room temperature. The mixture was then neutralized with an acidic Amberlite™ IR 120 H resin The resin was then removed by filtration and repeatedly extracted with methanol. The combined filtrate was concentrated under vacuum to give a crude product, which was recrystallized from MeOH to yield pure glycoside 4 (0.020 g, yield 50%) as a pale yellow solid: mp: 190–194 °C. [α]20D = −36.9 (c 0,17; MeOH). Rf = 0.15 (9:1 AcOEt–MeOH). 1H NMR (400 MHz; DMSO-d6): δ 8.15 (s, 1H; Ar), 8.06 (d, J = 9.0 Hz, 1H, NH), 7.82–7.91 (m, 3H; Ar), 7.50–7.55 (m, 2H; Ar), 7.44 (t, 1H, J = 7.3 Hz; Ar), 7.11 (s, 1H; Ar), 5.18–5.21 (m, 2H, 2 × OH), 5.11 (d, J = 8.7 Hz, 1H, anomeric CHO), 4.39 (t, J = 4.8 Hz, 1H, OH), 3.92 (s, 3H, COOMe), 3.80 (q, J = 9.0 Hz, 1H; CHO), 3.41–3.67 (m, 4H; CH2O, CHO and CHN), 3.11–3.19 (m, 1H; CHO), 1.94 (s, 3H; CH3CO); 13C NMR (100 MHz; DMSO-d6): δ 169.4, 159.4, 139.1, 137.7, 137.5, 129.9, 129.2 (2C), 128.1, 127.4 (2C), 124.7 (q, J = 271.0 Hz), 122.4 (q, J = 32.3 Hz), 119.4 (q, J = 4.7 Hz), 117.2 (q, J = 2.0 Hz), 113.7, 106.5, 104.2, 77.0, 73.5, 69.8, 60.8, 54.0, 52.3, 23.2. HRMS: (M + H+) found 539.1639; C25H26F3N2O8 requires 539.1641.
:1 hexane–AcOEt (0.1% Et3N) mixture afforded pure glycoside 16 (0.206 g, 77% yield) as a liquid. Rf = 0.33 (4:6 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 8.13–8.27 (m corresponding to two diastereoisomers, overall 1H; Ar), 7.62–7.96 (m, 3H; Ar), 7.15–7.52 (m, 4H; Ar), 6.23–6.47 (m corresponding to two diastereoisomers, overall 2H; vinyl CH), 5.96–6.05 (m corresponding to two diastereoisomers, overall 1H; anomeric CH), 4.31–4.59 (m corresponding to two diastereoisomers, overall 1H; THP–CHO2), 3.08–4.15 (m corresponding to two diastereoisomers, overall 6H; CHO & CH2O), 3.96 (s, 3H; COOCH3), 1.29–1.74 (m corresponding to two diastereoisomers, overall 6H; THP–CH2).:1 t-BuOH–acetone mixture (0.31 mL) was added to a 50% p/v aqueous solution of N-methyl morpholine-N-oxide (NMO) (80 μL) and the resulting reaction mixture was treated with 2.5% p/v OsO4 solution in t-BuOH (80 μL) and stirred for 2 h at the same temperature. Dilution with AcOEt and evaporation of the filtered (Celite®) organic solution afforded a crude reaction product (0.092 g), which was subjected to flash chromatography. Elution with a 2:8 hexane–AcOEt (0.1% Et3N) mixture afforded 18 (0.037 g, 70% yield), practically pure as a liquid. Rf = 0.12 (2:8 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 8.25–8.31 (m corresponding to two diastereoisomers, overall 1H; Ar), 7.58–7.79 (m, 3H; Ar), 7.34–7.54 (m, 4H; Ar), 5.37–5.49 (m corresponding to two diastereoisomers, overall 1H; anomeric CHO), 5.14–5.25 (m corresponding to two diastereoisomers, overall 1H; THP–CHO2), 3.90–4.46 (m corresponding to two diastereoisomers, overall 5H; CH2O and CHO), 3.98 (s, 3H; COOMe), 3.34–3.80 (m corresponding to two diastereoisomers, overall 3H; CH2O and CHO), 1.40–1.74 (m corresponding to two diastereoisomers, overall 6H; THP–CH2).:1 hexane–acetone). [α]20D: +57.71 (c 0.35, CH3OH). 1H NMR (400 MHz; CD3OD) δ 8.31–8.36 (m, 1H; Ar), 7.72–7.79 (m, 3H; Ar), 7.45–7.54 (m, 2H; Ar), 7.40 (tt, 1H, J = 7.4, 1.2 Hz; Ar), 7.23 (qd, 1H, J = 1.8, 0.9 Hz; Ar), 5.47 (d, 1H, J = 8.3 Hz; anomeric CHO), 4.11 (t, 1H, J = 3.4 Hz; CHO), 4.03 (dd, 1H, J = 8.3, 3.4 Hz; CHO), 3.98 (s, 3H; COOMe), 3.93 (td, 1H, J = 6.2, 1.1 Hz; CHO), 3.81–3.85 (m, 1H; CHO), 3.79 (dd, 1H, J = 10.8, 4.6 Hz; CH2O), 3.66 (dd, 1H, J = 10.8, 6.1 Hz; CH2O). 13C NMR (62.5 MHz; CD3OD) δ 162.2, 141.2, 140.1, 139.9, 130.1 (2C), 129.8, 129.1, 128.4 (2C), 125.9 (q, J = 271.7 Hz), 124.4 (q, J = 32.3 Hz), 120.1 (q, J = 4.6 Hz), 118.4 (q, J = 1.3 Hz), 115.2, 108.8, 106.7, 75.5, 73.2, 70.4, 68.6, 61.9, 52.4. HRMS: (M + H+) found 498.1370; C23H23F3NO8 requires 498.1376.:7 hexane–AcOEt (0.1% Et3N) mixture afforded pure trans diol 12 (0.235 g, 45% yield), as a colorless liquid. Rf = 0.13 (3:7 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 6.58–6.66 (m corresponding to two diastereoisomers, overall 1H, vinyl CHO), 4.95–5.05 (m corresponding to two diastereoisomers, overall 1H, vinyl CH), 4.64–4.72 (m corresponding to two diastereoisomers, overall 1H, THP–CHO2), 4.06–4.20 (m corresponding to two diastereoisomers, overall 1H; CHO), 3.77–4.04 (m corresponding to two diastereoisomers, overall 5H CHO and CH2O), 3.47–3.62 (m corresponding to two diastereoisomers, overall 1H; CHO), 1.69–1.85 (m corresponding to two diastereoisomers, overall 3H, THP–CH2), 1.48–1.68 (m corresponding to two diastereoisomers, overall 3H; THP–CH2).:2 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 6.49–6.58 (m corresponding to two diastereoisomers, overall 1H; vinyl CHO), 4.81–4.90 (m corresponding to two diastereoisomers, overall 1H; vinyl CH), 4.63–4.71 (m corresponding to two diastereoisomers, overall 1H; THP–CHO2), 3.95–4.17 (m corresponding to two diastereoisomers, overall 2H; CHO and CH2O), 3.72–3.94 (m corresponding to two diastereoisomers, overall 4H CHO and CH2O), 3.43–3.62 (m corresponding to two diastereoisomers, overall 1H, CHO), 1.44–1.86 (m corresponding to two diastereoisomers, overall 6H; THP–CH2), 0.87 (s, 9H; (CH3)3CSi), 0.10 (s, 6H; CH3Si).:2 hexane–AcOEt (0.1% Et3N) mixture afforded pure mesylate derivative 14 (0.254 g, 71% yield) as a yellow liquid. Rf = 0.19 (8:2 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 6.45–6.54 (m corresponding to two diastereoisomers, overall 1H; vinyl CHO), 4.82–4.91 (m corresponding to two diastereoisomers, overall 1H; vinyl CH), 4.58–4.76 (m corresponding to two diastereoisomers, overall 2H; CHOMs + THP–CHO2), 4.22–4.34 (m corresponding to two diastereoisomers, overall 1H; CHO), 4.15–4.21 (m corresponding to two diastereoisomers, overall 1H; CHO), 3.81–4.02 (m corresponding to two diastereoisomers, overall 2H; CH2O), 3.57–3.80 (m corresponding to two diastereoisomers, overall 1H; CHO), 3.43–3.56 (m corresponding to two diastereoisomers, overall 1H; CHO), 3.04–3.10 (m corresponding to two diastereoisomers, overall 3H; CH3SO2), 1.66–1.89 (m corresponding to two diastereoisomers, overall 2H; THP–CH2), 1.44–1.62 (m corresponding to two diastereoisomers, overall 4H; THP–CH2), 0.83–0.92 (m corresponding to two diastereoisomers, overall 9H; (CH3)3CSi), 0.09–0.16 (m corresponding to two diastereoisomers, overall 6H; CH3Si).:1 hexane–AcOEt) to produce pure trans-hydroxymesylate 9 (0.122 g, 67% yield) as a yellow liquid. Rf = 0.19 (1:1 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 6.53–6.61 (m corresponding to two diastereoisomers, overall 1H; vinyl CHO), 4.93–5.04 (m corresponding to two diastereoisomers, overall 1H; vinyl CH), 4.72–4.87 (m corresponding to two diastereoisomers, overall 1H; THP–CHO2), 4.59–4.68 (m corresponding to two diastereoisomers, overall 1H; CHOMs), 4.18–4.30 (m corresponding to two diastereoisomers, overall 2H; CH2O), 3.43–4.05 (m corresponding to two diastereoisomers, overall 4H CHO & CH2O), 3.04–3.13 (m corresponding to two diastereoisomers, overall 3H; CH3SO2), 1.36–1.90 (m corresponding to two diastereoisomers, overall 6H; THP–CH2).:1 hexane–AcOEt (0.1% Et3N) mixture afforded pure glycoside 17 (0.155 g, 58% yield) as a white solid. Rf = 0.33 (4:6 hexane–AcOEt); mp 50–53 °C; 1H NMR (250 MHz; CDCl3) δ 7.77–7.89 (m corresponding to two diastereoisomers, overall 1H; Ar), 7.59–7.76 (m corresponding to two diastereoisomers, overall 3H; Ar), 7.34–7.56 (m corresponding to two diastereoisomers, overall 3H; Ar), 7.27–7.33 (m corresponding to two diastereoisomers, overall 1H; Ar), 6.12–6.36 (m corresponding to two diastereoisomers, overall 2H, vinyl CH), 5.78–5.89 (m corresponding to two diastereoisomers, overall 1H; anomeric CHO), 3.26–4.53 (m corresponding to two diastereoisomers, overall 7H; CH2O and CHO), 3.94 (s, 3H; COOMe), 1.28–1.86 (m corresponding to two diastereoisomers, overall 6H; THP–CH2).:1 t-BuOH–acetone mixture (0.25 mL) was added to 50% p/v aqueous solution of N-methyl morpholine-N-oxide (NMO) (60 μL) and the resulting reaction mixture was treated with 2.5% p/v OsO4 solution in t-BuOH (60 μL) and stirred for 8 h at room temperature. Dilution with AcOEt and evaporation of the filtered (Celite®) organic solution afforded a crude reaction product which was purified by flash chromatography. Elution with a 2:8 hexane–AcOEt (0.1% Et3N) mixture afforded pure α-mannopyranoside 19 (0.028 g, 65% yield) as a white solid. Rf = 0.17 (2:8 hexane–AcOEt). 1H NMR (250 MHz; CDCl3) δ 7.76–7.88 (m corresponding to two diastereoisomers, overall 1H; Ar), 7.55–7.72 (m corresponding to two diastereoisomers, overall 3H; Ar), 7.32–7.50 (m corresponding to two diastereoisomers, overall 4H; Ar), 5.66–5.71 (m corresponding to two diastereoisomers, overall 1H; anomeric CHO), 4.66–4.77 (m corresponding to two diastereoisomers, overall 1H; THP–CHO2), 4.30–4.47 (m corresponding to two diastereoisomers, overall 2H; CHO), 3.93–4.22 (m corresponding to two diastereoisomers, overall 3H; CHO and CH2O), 3.90 (s, 3H; COOMe), 3.71–3.86 (m corresponding to two diastereoisomers, overall 2H; CH2O), 3.29–3.48 (m corresponding to two diastereoisomers, overall 1H; CHO), 1.30–1.71 (m corresponding to two diastereoisomers, overall 6H; THP–CH2).:1 AcOEt–acetone). [α]20D +18.14 (c 0.95, CH3OH). 1H NMR (400 MHz; CD3OD) δ 8.04 (s, 1H; Ar), 7.75–7.78 (m, 3H; Ar), 7.48–7.53 (m, 2H; Ar), 7.41 (tt, 1H, J = 7.4, 1.2 Hz; Ar), 7.25–7.29 (m, 1H; Ar), 5.57 (d, 1H, J = 1.9 Hz; anomeric CHO), 4.57–4.63 (m, 1H; CHO), 4.20–4.28 (m, 1H; CHO), 3.96 (s, 3H; COOMe), 3.83–3.89 (m, 4H; CH2O and CHO). 13C NMR (62.5 MHz; CD3OD) δ 161.3, 141.1, 140.5, 139.2, 130.2 (2C), 129.3, 129.1, 128.5 (2C), 125.2 (q, J = 271.0 Hz), 124.8 (q, J = 32.9 Hz), 120.2 (q, J = 4.9 Hz), 118.3 (q, J = 1.7 Hz), 113.6, 111.2, 106.9, 77.5, 72.4, 70.5, 67.9, 62.6, 52.8. HRMS: (M + H+) found 498.1366; C23H23F3NO8 requires 498.1376.:1 CHCl3/acetone mixture yielded pure compound 21 (0.140 g, yield 40%) as a pale yellow solid: mp 222–225 °C. [α]20D = +26.9 (c 0.79; CHCl3). Rf = 0.3 (9:1 CHCl3–acetone). 1H NMR (400 MHz; CDCl3): δ 8.10 (s, 1H; Ar), 7.74 (s, 1H; Ar), 7.63 (d, 2H, J = 7.7 Hz; Ar), 7.36–7.49 (m, 3H; Ar), 7.33 (bs, 1H; Ar), 6.70 (d, 1H, J = 8.6 Hz; NH), 5.52 (d, 1H, J = 9.05, anomeric CHO), 5.16–5.28 (m, 2H; CH2OAc), 4.55 (q, 1H, J = 9.0 Hz; CHOAc), 4.26 (dd, 1H, J = 12.4, 4.7 Hz; CHOAc), 3.98–4.08 (m, 1H; CHO), 3.95 (s, 3H; COOMe), 3.69–3.75 (m, 1H; CHN), 2.07 (s, 3H; CH3CO), 2.03 (s, 3H; CH3CO), 2.02 (s, 3H; CH3CO), 1.77 (s, 3H; CH3CO). 13C NMR (100 MHz; CDCl3): δ 171.0, 170.6 (2C), 169.4, 161.0, 140.2, 140.0, 139.6, 129.2 (2C), 128.3, 127.4 (2C), 124.3 (q, J = 272.4 Hz), 123.8 (q, J = 33.0 Hz), 120.0 (q, J = 4.8 Hz), 119.03 (q, J = 4.7 Hz), 117.4 (q, J = 2.1 Hz), 114.3, 108.4, 105.8, 73.6, 72.5, 68.0, 61.8, 52.5, 52.4, 23.3, 20.8, 20.7, 20.4.:3 mixture of CH2Cl2 and MeOH (4 mL) and cooled at 0 °C. A freshly prepared 0.33 M solution of MeONa/MeOH (0.05 mL) was added to the resulting solution and the reaction mixture was stirred for 4 h at room temperature. The mixture was then neutralized with an acidic Amberlite™ IR 120 H resin The resin was then removed by filtration and repeatedly extracted with methanol. The combined filtrate was concentrated under vacuum to give a crude product, which was recrystallized from MeOH to yield pure glycoside 4 (0.020 g, yield 50%) as a pale yellow solid: mp: 190–194 °C. [α]20D = −36.9 (c 0,17; MeOH). Rf = 0.15 (9:1 AcOEt–MeOH). 1H NMR (400 MHz; DMSO-d6): δ 8.15 (s, 1H; Ar), 8.06 (d, J = 9.0 Hz, 1H, NH), 7.82–7.91 (m, 3H; Ar), 7.50–7.55 (m, 2H; Ar), 7.44 (t, 1H, J = 7.3 Hz; Ar), 7.11 (s, 1H; Ar), 5.18–5.21 (m, 2H, 2 × OH), 5.11 (d, J = 8.7 Hz, 1H, anomeric CHO), 4.39 (t, J = 4.8 Hz, 1H, OH), 3.92 (s, 3H, COOMe), 3.80 (q, J = 9.0 Hz, 1H; CHO), 3.41–3.67 (m, 4H; CH2O, CHO and CHN), 3.11–3.19 (m, 1H; CHO), 1.94 (s, 3H; CH3CO); 13C NMR (100 MHz; DMSO-d6): δ 169.4, 159.4, 139.1, 137.7, 137.5, 129.9, 129.2 (2C), 128.1, 127.4 (2C), 124.7 (q, J = 271.0 Hz), 122.4 (q, J = 32.3 Hz), 119.4 (q, J = 4.7 Hz), 117.2 (q, J = 2.0 Hz), 113.7, 106.5, 104.2, 77.0, 73.5, 69.8, 60.8, 54.0, 52.3, 23.2. HRMS: (M + H+) found 539.1639; C25H26F3N2O8 requires 539.1641.Molecular modeling
The compounds were built using Maestro 9.0 (ref. 24) and was subjected to a conformational search (CS) of 1000 steps, using a water environment model (generalized-Born/surface-area model) by means of Macromodel.25 The algorithm used was based on the Monte Carlo method with the MMFFs force field and a distance-dependent dielectric constant of 1.0. The ligand was then energy minimized using the conjugated gradient (CG) method until a convergence value of 0.05 kcal (mol−1 Å−1) was reached, using the same force field and parameters used for the CS. The hLDH5 chain was extracted from the minimized average structure of the complex between LDH and 1J obtained by us through molecular dynamic simulations.10 Automated docking was carried out by means of the GOLD 5.1 program.26 The “allow early termination” option was deactivated, the remaining GOLD default parameters were used, and the ligand was submitted to 30 genetic algorithm runs by applying the ChemScore fitness function. The best docked conformation was taken into account. The so obtained complexes were energy minimized using AMBER 11.27 Each complex was placed in a rectangular parallelepiped water box, an explicit solvent model for water (TIP3P) was used, and the complex was solvated with a 10 Å water cap. Chloride ions were added as counterions to neutralize the system. Two steps of minimization were then carried out. In the first stage, we kept the complex fixed with a position restraint of 500 kcal (mol−1 Å−2) and we solely minimized the positions of the water molecules. In the second stage, we minimized the entire system through 20000 steps of steepest descent followed by conjugate gradient until a convergence of 0.05 kcal (mol−1 Å−1) was attained. All the α carbons of the protein were blocked with a harmonic force constant of 10 kcal (mol−1 Å−2). Ten nanoseconds of MD simulation were then carried out. The time step of the simulations was 2.0 fs with a cutoff of 10 Å for the non-bonded interaction, and SHAKE was employed to keep all bonds involving hydrogen atoms rigid. Constant-volume periodic boundary MD was carried out for 400 ps, during which the temperature was raised from 0 to 300 K. Then 9.6 ns of constant pressure periodic boundary MD was carried out at 300 K using the Langevin thermostat to maintain constant the temperature of our system. General Amber Force Field (GAFF) parameters were assigned to the ligand, while partial charges were calculated using the AM1-BCC method as implemented in the Antechamber suite of AMBER 11. The final structure of the complex was obtained as the average of the last 8 ns of MD minimized by the CG method until a convergence of 0.05 kcal (mol−1 Å−1). The average structure was obtained using the ptraj program implemented in AMBER 11.
Biological assays
Compounds were identified using AMDIS Chromatogram software (Amdis, freeware available from amdis.net) and programmed WIST and Niley commercial libraries. The integration area of lactate in each sample was divided by the integration area of CPA in the same sample to achieve a lactate/internal standard ratio. The ratios were averaged for duplicates, and percent lactate production over vehicle was calculated for each independent experiment. The mean lactate production/vehicle was then averaged between three or more independent experiments.
These LC-MS parameters allowed for the clear resolution of compound 1 (elution time: 12.4 minutes in the UV trace, 12.6 minutes in the TIC), compound 2 (elution time: 12.7 minutes in the UV trace, 12.9 minutes in the TIC), compound 3 (elution time: 12.9 minutes in the UV trace, 13.1 minutes in the TIC), and 15 (elution time: 15.8 minutes in the UV trace; 16.0 minutes in the TIC). The UV traces of vehicle-treated sonicates from both the start and end of the experiment time course contained no peaks in this range. Calibration curves of 15 and glycoconjugates 1, 2, and 3 demonstrated a linear relationship between concentration and UV trace integration area, so a linear equation was generated for each compound to convert integration area to concentration.
Acknowledgements
Support from the National Institutes of Health (R01GM098453) is gratefully acknowledged. ECC is supported by an NCI F30 Ruth L. Kirschstein National Research Service Award (1F30CA168323).Notes and references
- O. Warburg, Science, 1956, 124, 269–270 CAS.
- P. F. Oliveira, A. D. Martins, A. C. Moreira, C. Y. Cheng and M. G. Alves, Med. Res. Rev., 2015, 35, 126–151 CrossRef PubMed.
- C. Granchi and F. Minutolo, ChemMedChem, 2012, 7, 1318–1350 CrossRef CAS PubMed.
- C. Granchi, D. Fancelli and F. Minutolo, Bioorg. Med. Chem. Lett., 2014, 24, 4915–4925 CrossRef CAS PubMed.
- (a) V. R. Fantin, J. St-Pierre and P. Leder, Cancer Cell, 2006, 9, 425–434 CrossRef CAS PubMed; (b) H. Xie, J. Hanai, J. G. Ren, L. Kats, K. Burgess, P. Bhargava, S. Signoretti, J. Billiard, K. J. Duffy, A. Grant, X. Wang, P. K. Lorkiewicz, S. Schatzman, M. Bousamra II, A. N. Lane, R. M. Higashi, T. W. Fan, P. P. Pandolfi, V. P. Sukhatme and P. Seth, Cell Metab., 2014, 19, 795–809 CrossRef CAS PubMed.
- T. Kanno, K. Sudo, M. Maekawa, Y. Nishimura, M. Ukita and K. Fukutake, Clin. Chim. Acta, 1988, 173, 89–98 CrossRef CAS.
- C. Granchi, S. Bertini, M. Macchia and F. Minutolo, Curr. Med. Chem., 2010, 17, 672–697 CrossRef CAS.
- C. Granchi, I. Paterni, R. Rani and F. Minutolo, Future Med. Chem., 2013, 5, 1967–1991 CrossRef CAS PubMed.
- E. C. Calvaresi and P. J. Hergenrother, Chem. Sci., 2013, 4, 2319–2333 RSC.
- C. Granchi, S. Roy, C. Giacomelli, M. Macchia, T. Tuccinardi, A. Martinelli, M. Lanza, L. Betti, G. Giannaccini, A. Lucacchini, N. Funel, L. G. León, E. Giovannetti, G. J. Peters, R. Palchaudhuri, E. C. Calvaresi, P. J. Hergenrother and F. Minutolo, J. Med. Chem., 2011, 54, 1599–1612 CrossRef CAS PubMed.
- C. Granchi, E. C. Calvaresi, T. Tuccinardi, I. Paterni, M. Macchia, A. Martinelli, P. J. Hergenrother and F. Minutolo, Org. Biomol. Chem., 2013, 11, 6588–6596 CAS.
- E. C. Calvaresi, C. Granchi, T. Tuccinardi, V. Di Bussolo, R. W. Huigens, H. Y. Lee, R. Palchaudhuri, M. Macchia, A. Martinelli, F. Minutolo and P. J. Hergenrother, ChemBioChem, 2013, 14, 2263–2267 CrossRef CAS PubMed.
- J. E. Barnett, G. D. Holman and K. A. Munday, Biochem. J., 1973, 131, 211–221 CAS.
- H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore and J. Darnell, in Molecular Cell Biology, ed. W. H. Freeman, New York, 4th edn, 2000, http://www.ncbi.nlm.nih.gov/books/NBK21669/ Search PubMed.
- Z. Ma and K. Vosseller, Amino Acids, 2013, 45, 719–733 CrossRef CAS PubMed.
- Z. Ma and K. Vosseller, J. Biol. Chem., 2014, 289, 34457–34465 CrossRef PubMed.
- V. Di Bussolo, M. Caselli, M. Pineschi and P. Crotti, Org. Lett., 2002, 4, 3695–3698 CrossRef CAS PubMed.
- V. Di Bussolo, M. Caselli, M. Pineschi and P. Crotti, Org. Lett., 2003, 5, 2173–2176 CrossRef CAS PubMed.
- V. Di Bussolo, M. Caselli, M. R. Romano, M. Pineschi and P. Crotti, J. Org. Chem., 2004, 69, 7383–7386 CrossRef CAS PubMed.
- V. Di Bussolo, L. Checchia, M. R. Romano, M. Pineschi and P. Crotti, Org. Lett., 2008, 10, 2493–2496 CrossRef CAS PubMed.
- S. Nakabayashi, C. D. Warren and R. W. Jeanlozs, Carbohydr. Res., 1986, 150, C7–C10 CrossRef CAS.
- K. Křenek, P. Šimon, L. Weignerová, B. Fliedrová, M. Kuzma and V. Křen, Beilstein J. Org. Chem., 2012, 8, 428–432 CrossRef PubMed.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed.
- Maestro, version 9.0, Schrödinger Inc, Portland, OR, 2009 Search PubMed.
- Macromodel ver. 9.7, Schrödinger Inc, Portland, OR, 2009 Search PubMed.
- M. L. Verdonk, J. C. Cole, M. J. Hartshorn, C. W. Murray and R. D. Taylor, Proteins, 2003, 52, 609–623 CrossRef CAS PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 11, University of California, San Francisco, CA, 2010 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of 2–4, 9, 12–14, 16–19 and 21; HRMS of 2–4; calibration curves of 1–3. See DOI: 10.1039/c5ra00946d |
| ‡ Present address: College of Medicine, University of Illinois, 506 S. Mathews Avenue, Urbana, IL 61801, USA. |
| § Present address: Dipartimento di Biotecnologia, Chimica e Farmacia, Università degli Studi di Siena, via Aldo Moro 2, 53019 Siena, Italy. |
| This journal is © The Royal Society of Chemistry 2015 |