
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tunable band gap and coercivity of bismuth ferrite–polyaniline core–shell nanoparticles: the role of shell thickness
Smita
Chaturvedi
*a,
Raja
Das
bcd,
Pankaj
Poddar
bc and
Sulabha
Kulkarni
a
aIndian Institute of Science Education and Research Pune, Dr Homi Bhabha Road, Pune-411008, India. E-mail: smita.chaturvedi24@gmail.com
bPhysical and Materials Chemistry Division, CSIR-National Chemical Laboratory, Pashan, Pune-411008, India
cAcademy of Scientific and Innovative Research (AcSIR), Anusandhan Bhavan, 2 Rafi Marg, New Delhi-100001, India
dDepartment of Physics, University of South Florida, 4202 East Fowler Ave, PHY114, Tampa, FL 33620, USA
First published on 3rd March 2015
Abstract
We report a tunable band gap of bismuth ferrite–polyaniline core–shell nanoparticles from 2.24 to 1.98 eV and the variation of coercivity from 118 to 100 Oe, by varying the thickness of the polyaniline shell. Bismuth ferrite nanoparticles were synthesized via a chemical route and coating of polyaniline was achieved by a double surfactant layer technique. Coating of polyaniline introduced oxygen vacancies, local distortions and microstrains in the bismuth ferrite lattice which resulted in a change in the Fe–O–Fe bond angle of the bismuth ferrite lattice. This is confirmed with structural analysis tools i.e. X-ray Diffraction, Fourier Transform Infrared Spectroscopy and Raman spectroscopy. Moreover, the absorption of light around 600 nm is increased and photoluminescence emission around 430 nm is decreased, as the thickness of shell is increased, which also indicates the changes in optical properties due to variation in shell thickness. Thus tunability of the band gap and coercivity is achieved as a function of shell thickness in bismuth ferrite–polyaniline core–shell nanoparticles.
1. Introduction
Bismuth ferrite, BiFeO3 (BFO) is a widely investigated multiferroic material which has a very high ferroelectric Curie temperature (TC ∼ 1100 K) and antiferromagnetic Nèel temperature (TN ∼ 643 K). In the nano size regime BFO particles inherently assume a core–shell structure in which the core is antiferromagnetic and the shell is ferromagnetic. The ferromagnetic shell thickness increases with decreasing particle size.1 Additional coatings like silica, polyaniline and NiFe2O4 showed remarkable changes in the magnetic properties of the BFO nanoparticles.2–4 Liu et al. have very recently reported controllable synthesis and enhanced photocatalytic activity of BFO–C core–shell nanofibres and showed that introduction of carbon should not only enhance the light absorption of BFO nanoparticles and adsorption capacity of methyl orange but also facilitate the separation of photo-generated electron–hole pair, both of which result in enhanced photocatalytic activity of BFO nanofibers.5 Thus by coating the BFO nanoparticles indeed it is possible to influence its properties.Multiferroic materials with small band gaps are potential candidates for optoelectronics and device applications.6 Until recent past BFO has been known and investigated for its multiferroic characteristics and applications. Currently, due to narrow band gap and p-type semiconductor characteristics, BFO is also being considered a potential candidate for photocatalysis as well as photovoltaics.7,8 Nanoparticles of BFO in the size range 5 nm to 65 nm were shown to have variation in the band gap from 2.32 to 2.09 eV.9 The band gap variation was attributed to the strain and oxygen vacancies in the nanoparticles. In this work we demonstrate that polyaniline (PANI) coating thickness can influence the optical band gap of BFO nanoparticles. To the best of our knowledge, a multiferroic-polymer core–shell combination for tuning the band gap and coercivity has not been explored so far. Here we achieve a variation in the band gap of BFO from 2.24 eV to 1.98 eV and changes in the coercivity from ∼118 to ∼100 Oe by increasing the polyaniline shell thickness from 0 nm to 15 nm.
2. Experimental
2.1. Synthesis of BFO nanoparticles
BFO nanoparticles were synthesized using a soft chemical route combined with post synthesis annealing, reported earlier.3 Briefly, it involves a reaction of Bi(NO3)3·5H2O and Fe(NO3)3·9H2O in the presence of tartaric acid at 353 K. The precipitate is heated in an oven at 423 K. After washing in dilute nitric acid (∼17%) and ethanol, it is annealed at 873 K for 2 h. The annealed powder is washed in Milli-Q water and ethanol several times, and then completely dried. The sample is simply referred to as BFO nanoparticles.2.2. Synthesis of BFO–PANI core–shell nanoparticles
Coating of conducting polymer on multiferroic nanoparticles is interesting because the surface of nanoparticles can be tailored to control specific magnetic, electric, optical, optoelectronic and catalytic properties. Also polyaniline is one of the most important conducting polymers with high conductivity, ease of synthesis, low cost, environmental and chemical stability, and unique redox behavior.10,11 The synthesis of BFO–PANI core–shell NPs is challenging as it requires generating the polymer coating uniformly and completely on the surface of the metal oxide core by a polymerization reaction in a solution phase. The difficulty is at slowing down the rate of polymerization and controlling the polymerization on the surface of the core rather than in the solution. In the process of coating of BFO with PANI, the requirement of high acidic environment for polymerization of PANI leads to etching of the BFO core of the material. To overcome this problem we have adapted synthetic route called double-surfactant-layer (DSL)-assisted polymerization method,3,12 which effectively controls the polymerization of PANI onto the surface of BFO.To improve the stability of the BFO core, the resultant BFO particles were redispersed in a solution composed of 120 ml Milli-Q water and 1 g polyvinylpyrrolidone (PVP: MW 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). The mixture was ultrasonicated for 12 h to ensure that the adsorption of PVP was complete. To remove unadsorbed PVP molecules, the dispersion was centrifuged and washed with ethanol several times. Resultant PVP stabilized BFO was redispersed in a solution composed of 50 g distilled water, 0.08 g sodium dodecyl sulphate (SDS), and 0.01 g (NH4)2S2O8 (APS). This solution was held for 12 h at room temperature. Then appropriate amount of aniline and 1.4 × 10−3 M HCl were added to the above mixture solution and the reaction was held at 277 K for 12 h with stirring. The resulting precipitate was centrifuged and washed several times with water and ethanol. To obtain different thicknesses of polyaniline coating on the surface of BFO, different concentrations of aniline were used i.e. 8 μL, 10 μL and 12 μL were used and prepared samples were named as BP1, BP2 and BP3 respectively. Hereafter, throughout the discussion, the samples, BFO and BFO–PANI core–shell are named as BFO and BP1, BP2, BP3 respectively.
000). The mixture was ultrasonicated for 12 h to ensure that the adsorption of PVP was complete. To remove unadsorbed PVP molecules, the dispersion was centrifuged and washed with ethanol several times. Resultant PVP stabilized BFO was redispersed in a solution composed of 50 g distilled water, 0.08 g sodium dodecyl sulphate (SDS), and 0.01 g (NH4)2S2O8 (APS). This solution was held for 12 h at room temperature. Then appropriate amount of aniline and 1.4 × 10−3 M HCl were added to the above mixture solution and the reaction was held at 277 K for 12 h with stirring. The resulting precipitate was centrifuged and washed several times with water and ethanol. To obtain different thicknesses of polyaniline coating on the surface of BFO, different concentrations of aniline were used i.e. 8 μL, 10 μL and 12 μL were used and prepared samples were named as BP1, BP2 and BP3 respectively. Hereafter, throughout the discussion, the samples, BFO and BFO–PANI core–shell are named as BFO and BP1, BP2, BP3 respectively.
2.3. Characterization
X-ray powder diffraction patterns (XRD) of the products were obtained using a Bruker D8 Advance X-ray diffractometer equipped with graphite monochromatized Cu K-α radiation (λ = 1.54178 Å). Transmission electron microscopy (TEM) photographs were taken on a FEI model Tecnai T20 G2 with LaB6 electron source to image BFO particles and BFO–PANI core–shell at an accelerating voltage of 200 kV. For recording TEM, samples were dispersed in ethanol, sonicated for 10 minutes and then drop casted on a copper TEM grid for imaging. Infrared (IR) spectra were recorded in the wavenumbers ranging from 4000 to 500 cm−1 with Thermo scientific NICOLET 6700 Fourier transform infrared (FT-IR) spectrometer. Raman spectroscopy was carried out using HR 800 Raman spectrophotometer (Jobin Yvon, Horiba, France) using monochromatic radiation (488 nm) operating at 20 mW. For recording Raman spectra, powder sample were spread evenly on a quartz glass plate and then exposed to incident laser. The magnetic measurements were performed using a Physical Property Measurement System (PPMS) (Quantum Design Inc., San Diego, California equipped with a 9 Tesla superconducting magnet). We collected M–H loops at a rate of 50 Oe s−1 in a field sweep from −85 to 85 kOe at the vibrating frequency of 40 Hz. UV spectra were obtained on Perkin Elmer Lambda 950 UV-Vis-NIR spectrometer using dilute solution of the samples in ethanol. For recording PL spectra, the samples were dispersed in ethanol. PL spectra were measured on Horiba Scientific Fluoromax-4 spectrofluorometer using 380 nm excitation wavelength.3. Results and discussion
3.1. Morphology and phase structure
Fig. 1(a)–(d) show the TEM images of BFO and BFO–PANI NPs respectively. | ||
| Fig. 1 TEM micrographs of BFO (a), BP1 (b), BP2 (c) and BP3 (d). | ||
The average size of BFO NPs was ∼85 nm and thicknesses of the shell were measured ∼5, 10 and 15 nm for BP1, BP2 and BP3 respectively. Fig. 2 shows the XRD patterns of BFO, BP1, BP2 and BP3. The patterns clearly indicate good quality crystallinity of BFO. It was found that the BFO nanoparticles as well as BP nanoparticles are in the rhombohedral structure and data is in good in agreement with JCPDS Card no. 01-072-7678 and is also in agreement with previously reported BFO nanoparticles.3,13,14 No other characteristic peaks due to any impurities were detected. The size of BFO particles determined using Scherer formula, d = 0.9λ/βcosθ is ∼80 nm, in close agreement with that observed by TEM measurements (here d = particle diameter, λ = wavelength of X rays used (1.54 Å), β = FWHM and θ is the Bragg angle). For the XRD of BP1, BP2 and BP3, all the peaks are similar to that of BFO, which indicates that the crystal structure of BFO is well maintained after the coating process despite acidic conditions. Probably because of the relatively thin layer of PANI, broad diffraction peak expected for amorphous PANI or any other impurity is not observed. The average size of BP1, BP2 and BP3 particles determined using Scherrer formula is ∼80 nm which indicates that size of BFO is maintained after coating. Fig. 2 also shows the magnification of partial 2θ range from 31 to 33 degrees, which contains characteristic splitting of rhombohedral peak. The doublet near 32° remains same for all the samples and broadening is observed which can be attributed to decreasing values of lattice parameters due to incorporation of PANI3 and it indicates that PANI might have formed the coordination with lattice oxygen, which has two major effects on BFO lattice (i) locally distort the crystal lattice of BFO by incorporation of PANI. Distortion of the local lattice is significant factor for supporting the changes observed in optical properties. (ii) Disturb the overall charge compensation of the BFO lattice. In turn it may generate some lattice defects, decreasing the lattice parameters.3 By incorporating PANI onto BFO, the bond angle of O–Bi–O and Fe–O–Fe increases. This brings Fe–Fe ions closer and contributes towards the changes in magnetic properties.3
 | ||
| Fig. 2 XRD spectra of BFO, BP1, BP2 and BP3 samples. | ||
The successful polymerization of aniline onto the surface of BFO was confirmed by Fourier Transform Infrared Spectroscopy (FTIR), as shown in Fig. 3. The characteristic peak of BFO appeared at 550 cm−1 in all the samples and the intensity is reducing as the thickness of shell is increasing. The characteristic peaks of PANI appeared at 1586, 1438 and 1308 cm−1, due to quinone ring deformation, benzene ring deformation and C–N stretching of a secondary aromatic amine, respectively which are comparable as reported by Huang et al.15 The expanded spectra around 1500 cm−1 is shown in Fig. 2(b). The band at 1070 cm−1 was ascribed to the adsorption of the –SO3H group, suggesting that the PANI shell is also doped with a little SDS.16 Results from FTIR measurements have provided evidence that the polymerization of PANI has been successfully obtained on the surface of the BFO NPs. Various characterization peaks of BFO, BP1, BP2 and BP3 and their assignments are shown in Table 1.
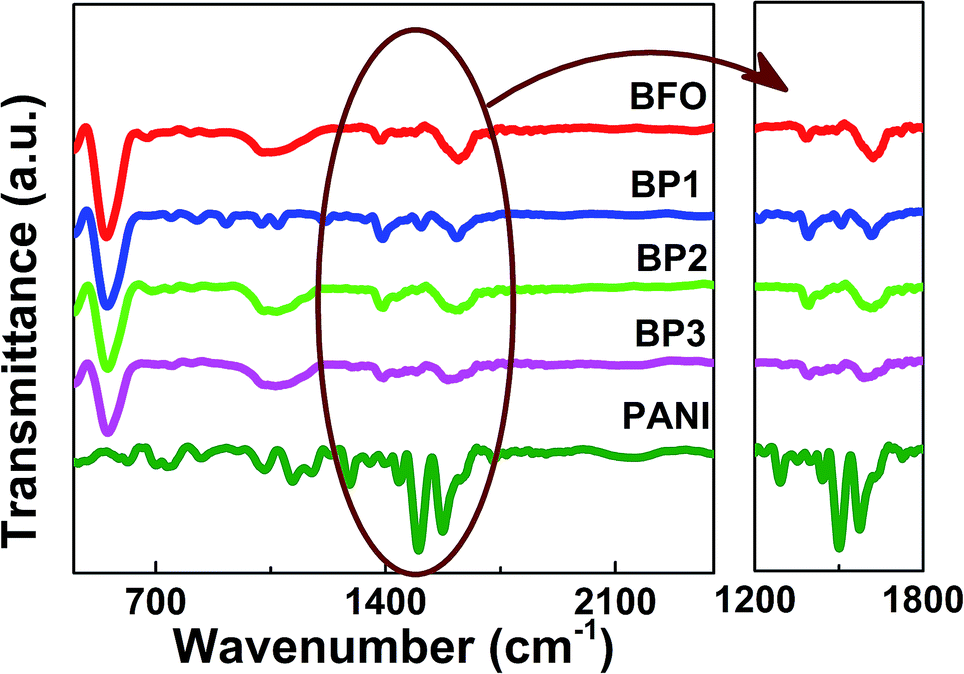 | ||
| Fig. 3 FTIR spectra of PANI, BFO, BP1, BP2 and BP3 samples. | ||
| Transmittance peaks (cm−1) | Assignment | ||||
|---|---|---|---|---|---|
| BFO | BP1 | BP2 | BP3 | PANI | |
| 550 | 550 | 550 | 550 | — | Fe–O stretching (BFO) |
| 1061 | 1061 | 1061 | 1061 | 1020 | Absorption of the –SO3H |
| — | 1304 | — | 1221 | 1291 | C–N stretching (PANI) |
| — | 1438 | 1494 | 1488 | 1507 | Benzene ring deformation (PANI) |
| 1628 | 1578 | 1608 | 1590 | 1615 | Quinone ring deformation (PANI) |
3.2. Raman analysis
The incorporation of PANI coating indicated by XRD analysis is supported by the Raman spectra analysis of BFO and BP samples. Raman spectroscopy in general is very sensitive to the structural changes. The Raman active modes of the rhombohedral BFO with R3c structure are summarized using the irreducible representation ΓRaman,R3c = 4A1 + 9E.17The Raman spectrum of BFO obtained using 488 nm excitation wavelength depicted in 4 (a) undergoes considerable changes after PANI coating which is shown in Fig. 4(b)–(d). The spectra have been deconvoluted into 10 individual components for BFO (3 A modes and 7 E modes), 7 components for BP1 (3 A modes and 4 E modes) and 8 components for BP2 and BP3 (3 A modes, 5 E modes and one mode from PANI). The deconvoluted Raman modes are listed in Table 2. For the sake of comparison the Raman modes for BFO single crystals and nanoparticles from the literature are also given.3,18 It can be observed that there are no impurity related peaks in BFO or BP sample supporting our XRD data. The overall spectral signature of BFO is comparable with the reported data for single crystal.19
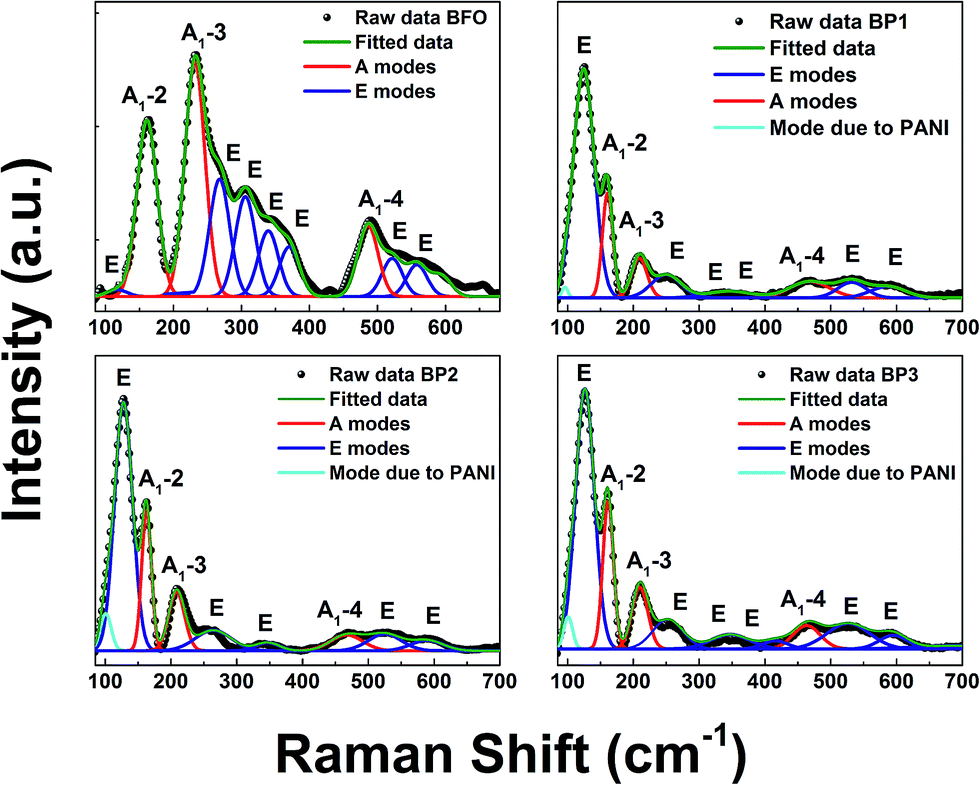 | ||
| Fig. 4 Raman spectra of BFO, BP1, BP2 and BP3 samples. | ||
| Raman modes | Ref. 17 | Present study (NPs) | |||
|---|---|---|---|---|---|
| BFO | BFO | BP1 | BP2 | BP3 | |
| A1-1 | 147 | — | — | — | — |
| A1-2 | 176 | 163 | 159 | 160 | 163 |
| A1-3 | 227 | 235 | 209 | 207 | 209 |
| A1-4 | 490 | 485 | 466 | 468 | 463 |
| E | 265 | 268 | 251 | — | 253 |
| E | 279 | 307 | 347 | 346 | 346 |
| E | 351 | 339 | — | — | 417 |
| E | 375 | 371 | — | — | — |
| E | 437 | — | — | — | — |
| E | 473 | 521 | 531 | 525 | 526 |
| E | 525 | 557 | 592 | 592 | 596 |
| E | 77 | — | — | — | — |
| E | 136 | 120 | 124 | 128 | 126 |
| Mode due to PANI | 95 | 100 | 100 | ||
The first principle calculation suggests that in BFO lattice, low frequency modes below 167 cm−1 are contributed due to Bi atoms, Fe atoms mainly participate in the modes between 152 and 262 cm−1, while for higher frequency modes above 262 cm−1, modes from oxygen atoms dominate.17,20 It is known that Raman spectra are sensitive to atomic displacements. Hence, the changes in the Raman modes due to incorporation of PANI onto the BFO lattice can provide significant information. Comparing BFO with BP1, BP2, BP3, it is observed that (i) the intensity of A1-2, A1-3 and A1-4 is decreased and the peaks are shifted as well which is due to increase in Fe–O–Fe bond angle, which places Fe–Fe atoms closer. This shifting and broadening of Raman modes in BP suggests the presence of disorder and the internal microstrain due to Fe ions and contributes to the changes observed in magnetic properties of the system. (ii) Further, it is known that sample preparation methods influence the oxygen stoichiometry. This is expected to show changes in oxygen bonding and disorder that is reflected in the vibrational frequencies of mode involving oxygen. Intensities of all the observed E modes above 262 cm−1 are decreased as well as the modes are broadening and shifting is noticed. This indicates that oxygen vacancies are created in the system, which distort the lattice and contributed to microstrain and local distortion of the BFO lattice. This explains the change in optical properties.
The extra mode centered around 100 cm−1 represents a mode due to PANI coating.21 Also it is observed that intensity of the mode due to PANI (around 100 cm−1) is increasing as the thickness of PANI layer is increased.
3.3. Magnetic properties: tuning coercivity
To investigate the magnetic behavior of BFO and BFO–PANI core–shell NPs, the magnetic measurements were recorded at room temperature. Fig. 5(a) and (b) show room temperature M–H loop and expanded plot of M–H loops. It is observed that magnetization as a function of applied field decreases as the thickness of the shell increases. This is due to reduction of net weight of BFO without PANI. Also, there is variation in value of negative exchange bias and coercivity as the thickness of PANI shell increases, as shown in Fig. 5(c), the values of negative exchange bias obtained for BFO, BP1, BP2 and BP3 were 22, 25, 23 and 24 Oe respectively. Coercivity for samples BFO, BP1, BP2 and BP3 were 118, 114, 104 and 100 Oe respectively. Fig. 5(d) shows linear dependence of magnetization on 1/d and this indicate that the simple Néel model is applicable.1 Hence, these changes in coercivity and exchange bias are attributed to following respectively, (i) microstrain as a result of oxygen vacancies and lattice defects introduced due to incorporation of PANI layer3 (ii) existence of antiferromagnetic (AFM) core and ferromagnetic (FM) shell within the BFO NPs provides possibility for exchange coupling between ferromagnetic surfaces and antiferromagnetic core1,22 and AFM-FM-PANI in core shell NPs.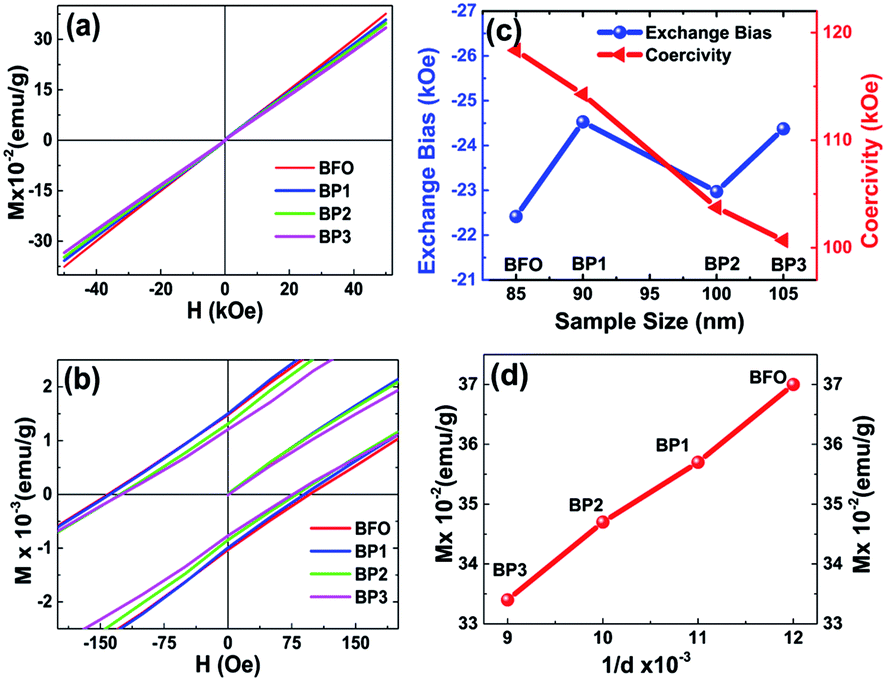 | ||
| Fig. 5 Room temperature hysteresis loops for BFO, BP1, BP2, BP3 (a), expanded view of the hysteresis loops (b), plot of exchange bias and coercivity as a function of shell thickness (c) and plot of 1/d and magnetization as a function of shell thickness (d). | ||
Since BFO NPs are coated with nonmagnetic PANI layer, which is limiting the interparticle interactions as well as cross grain boundary interactions. Hence, as the thickness of PANI coating increases, the interactions between particles and cross grain boundaries are further limited resulting in more decreased values of coercivity.
It is demonstrated that the PANI coating onto BFO effectively modifies Fe–O bond length (decreases) and Fe–O–Fe bond angle (increases).3 The decrease in Fe–O bond length enhances the Fe(3d)–O(2p) orbital overlap and decrease in Fe–O–Fe bond opposes the Fe(3d)–O(2p) orbital overlap. The extent of net orbital overlap determines the superexchange interactions between the neighboring Fe spins. As the thickness of PANI increases the amount of net orbital overlap changes and hence the magnetic properties, this is in agreement with XRD. Raman study of the core–shell NPs also supports aforementioned hypothesis that due to PANI coating the relative intensity of A1-2 mode is shifted which controls the Fe–O bond parameter.17,20
3.4. Optical properties: tuning band gap
It is observed that Band gap of BFO–PANI core–shell NPs is decreasing as we increase the shell thickness as illustrated in Fig. 6. | ||
| Fig. 6 Band gap of BFO–PANI core–shell NPs as a function of polyaniline shell thickness. | ||
Fig. 7(a) shows the UV-Vis absorption spectra of BFO, BP1, BP2 and BP3 at pH 12. The absorbance plot shows a strong transition in 500–600 nm range which corresponds to electronic transitions involving charge transfer from valence band O 2p states to conduction band Fe 3d states.9 Due to this transition the absorption edge is shifting towards the higher wavelength side as the thickness of PANI shell increases. Absorbance parameters of the BFO–PANI core shell can be used to calculate the electronic band gap of the BFO–PANI structures. When quantifying the band gap of the core–shell samples of different thicknesses of the PANI with UV-visible data, the electronic band structures are linked to emeraldine salt form of PANI (Eg = 1.5 eV).23
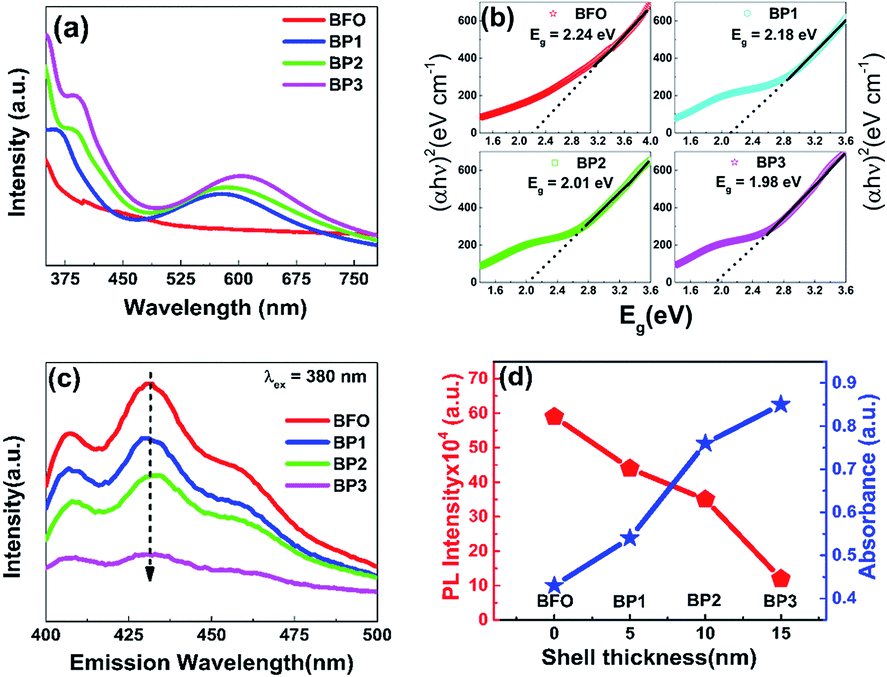 | ||
| Fig. 7 UV-Vis absorption spectra of BFO, BP1, BP2, BP3 and PANI (a), PL spectra for samples BFO, BP1, BP2 and BP3 (b), Tauc plot for band gap determination of samples BFO, BP1, BP2 and BP3 (c) and plot of PL intensity and absorbance as a function of shell thickness (d). | ||
The energy band gap of these NPs has been calculated with the help of absorption spectra (Fig. 7(a)). To calculate the optical band gap energy from absorption Spectra, the Tauc relation is used.24
| αhν = A(hν − Eg)n | (1) |
The values of band gap obtained are 2.24 eV, 2.18 eV, 2.01 eV and 1.98 eV for samples BFO, BP1, BP2 and BP3 respectively. The obtained value of band gap of BFO is in agreement with previously reported values.5,24 The decrease in band gap values has following important contributing factors:
(i) In BFO–PANI core–shell, PANI is in emeraldine salt form. Emeraldine salt form is conductive and has the work function ∼4.1–4.5 eV,25 which is comparable to the work function of silver (4.26 eV).26 Thus BFO–PANI core–shell NPs can be considered as p type semiconductor (BFO)–metal (PANI) composites. (ii) Further Room temperature PL emission spectra (excitation wavelength 380 nm) of BFO and BP1, BP2 and BP3 as shown in Fig. 7(c) also support the fact that the rate of recombination of electron hole pair is decreased, as PL spectrum of BFO shows a strong emission indicating high electron hole pair recombination rate, while PL spectra of BP1, BP2 and BP3 show remarkable decreased emission, confirming slow rate of electron hole pair recombination. On increasing the thickness of the PANI shell, PL emission decreases and absorbance increases, as shown in Fig. 7(d). (iii) Existence of defect induced energy levels between the conduction and valence band affects band-gap. The presence of microstrain and lattice parameter variation represent lattice distortion, which can be generated from the grain boundaries present in core shell NPs. This microstrain in the BFO–PANI core shell NPs modifies the energy levels, thereby influencing the absorption band edge. Moreover, microstrain in BFO are generated due to inherent oxygen vacancies. These oxygen vacancies are lower than the adjacent Fe 3d levels, resulting in the sub band-gap defect states.27 Further as discussed above that Fe(3d)–O(2p) orbital overlap varies as a function of shell thickness and hence further contributing to microstrain in the system and in turn decreased value of band gap. Also unsaturated bonds on the surface of the BFO–PANI core–shell NPs are also responsible for introducing microstrain, which create deep and shallow levels within the band gap.9
4. Summary and conclusions
In summary, BFO nanoparticles of ∼85 nm were synthesized. These BFO nanoparticles were coated with different thicknesses of polyaniline (∼5, 10 and 15 nm). A detailed study on the structural, magnetic, redox and optical properties of the BFO–PANI core shell NPS was conducted. The coercivity of the BFO–PANI core–shell is reduced by about 20 Oe for shell thickness of 15 nm. This tunable coercivity is a result of change of net superexchange in the system due to change in thickness of PANI shell. Further limited interparticle as well as cross grain boundary interactions also contribute to the tunability of coercivity and exchange bias. Tunable magnetization by exploiting the thickness effect of these nanoparticles is significant for specific applications at room temperature, involving spintronics and spin valves. It is observed that the tunable band gap from 2.2 eV to 1.98 eV is a function of PANI shell thickness (0 nm to 15 nm), suggesting that microstrain, oxygen defects and exchange interactions have a strong impact in determining the band gap values of NPs. This work places BFO–PANI NPs in class of potential material for simultaneous application in spintronics, photocatalysis, photovoltaic and optoelectronics.Acknowledgements
This work was carried out under the Department of Science and Technology, Ministry of Science and Technology Grant no. SR/WOS/-A/PS50/2012(G), India. S. K. thanks DST India Nano-Mission Initiative project SR/NM/NS-42/2009. The authors thank Neeraj Maheshwari for the X-ray Diffraction measurements, Sayan Mondal for the Raman spectroscopy measurements and Amey Apte and Prashant Bhaskar for the schematic illustration.Notes and references
- T. Park, G. C. Papaefthymiou, A. J. Viescas, A. R. Moodenbaugh and S. S. Wong, Nano Lett., 2007, 7, 766–772 CrossRef CAS PubMed.
- M. M. Shirolkar, R. Das, T. Maity, P. Poddar and S. K. Kulkarni, J. Phys. Chem. C, 2012, 116, 19503–19511 CAS.
- S. Chaturvedi, M. M. Shirolkar, R. Rajendra, S. Singh, N. Ballav and S. Kulkarni, J. Appl. Phys., 2014, 115, 123906 CrossRef.
- S. M. Yusuf, P. K. Manna, M. M. Shirolkar, S. K. Kulkarni, R. Tewari and G. K. Dey, J. Appl. Phys., 2013, 113, 173906 CrossRef.
- Y. Liu, R. Zuo and S. Qi, J. Mol. Catal. A: Chem., 2013, 376, 1–6 CrossRef CAS.
- A. Kumar, R. C. Rai, N. J. Podraza, S. Denev, M. Ramirez, Y.-H. Chu, L. W. Martin, J. Ihlefeld, T. Heeg, J. Schubert, D. G. Schlom, J. Orenstein, R. Ramesh, R. W. Collins, J. L. Musfeldt and V. Gopalan, Appl. Phys. Lett., 2008, 92, 121915 CrossRef.
- F. Gao, X. Y. Chen, K. B. Yin, S. Dong, Z. F. Ren, F. Yuan, T. Yu, Z. G. Zou and J.-M. Liu, Adv. Mater., 2007, 19, 2889–2892 CrossRef CAS.
- S. Y. Yang, L. W. Martin, S. J. Byrnes, T. E. Conry, S. R. Basu, D. Paran, L. Reichertz, J. Ihlefeld, C. Adamo, A. Melville, Y.-H. Chu, C.-H. Yang, J. L. Musfeldt, D. G. Schlom, J. W. Ager and R. Ramesh, Appl. Phys. Lett., 2009, 95, 062909 CrossRef.
- P. S. V. Mocherla, C. Karthik, R. Ubic, M. S. Ramachandra Rao and C. Sudakar, Appl. Phys. Lett., 2013, 103, 022910 CrossRef.
- L. Zhang and M. Wan, J. Phys. Chem. B, 2003, 107, 6748–6753 CrossRef CAS.
- L. Liang, J. Liu, C. F. Windisch Jr, G. J. Exarhos and Y. Lin, Angew. Chem., Int. Ed., 2002, 41, 3665–3668 CrossRef CAS.
- C.-L. Zhu, S.-W. Chou, S.-F. He, W.-N. Liao and C.-C. Chen, Nanotechnology, 2007, 18, 275604 CrossRef.
- S. Chaturvedi, I. Sarkar, M. M. Shirolkar, U.-S. Jeng, Y.-Q. Yeh, R. Rajendra, N. Ballav and S. Kulkarni, Appl. Phys. Lett., 2014, 105, 102910 CrossRef.
- L. Shen, P. E. Laibinis and T. A. Hatton, Langmuir, 1999, 15, 447–453 CrossRef CAS.
- K. Huang and M. Wan, Chem. Mater., 2002, 14, 3486–3492 CrossRef CAS.
- K. G. Neoh, M. Y. Pun, E. T. Kang and K. L. Tan, Synth. Met., 1995, 73, 209–215 CrossRef CAS.
- M. K. Singh, H. M. Jang, S. Ryu and M.-H. Jo, Appl. Phys. Lett., 2006, 88, 42907 CrossRef.
- H. Fukumura, H. Harima, K. Kisoda, M. Tamada, Y. Noguchi and M. Miyayama, J. Magn. Magn. Mater., 2007, 310, e367–e369 CrossRef CAS.
- R. Haumont, J. Kreisel, P. Bouvier and F. Hippert, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 132101 CrossRef.
- P. Hermet, M. Goffinet, J. Kreisel and P. Ghosez, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 220102 CrossRef.
- M. Baibarac, I. Baltog and S. Lefrant, J. Solid State Chem., 2009, 182, 827–835 CrossRef CAS.
- A. Jaiswal, R. Das, K. Vivekanand, P. Mary Abraham, S. Adyanthaya and P. Poddar, J. Phys. Chem. C, 2010, 114, 2108–2115 CAS.
- W. S. Huang and A. G. MacDiarmid, Polymer, 1993, 34, 1833–1845 CrossRef CAS.
- S. Mohan, B. Subramanian and G. Sarveswaran, J. Mater. Chem. C, 2014, 2, 6835 RSC.
- R. Gangopadhyay and A. De, Chem. Mater., 2000, 12, 608–622 CrossRef CAS.
- X. Zhang, Y. L. Chen, R.-S. Liu and D. P. Tsai, Rep. Prog. Phys., 2013, 76, 046401 CrossRef PubMed.
- A. J. Hauser, J. Zhang, L. Mier, R. A. Ricciardo, P. M. Woodward, T. L. Gustafson, L. J. Brillson and F. Y. Yang, Appl. Phys. Lett., 2008, 92, 222901 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |