
Controlling the coassembly of highly amphiphilic block copolymers with a hydrolytic sol by solvent exchange†
Stefan Guldin‡
 a,
Morgan Stefikb,
Hiroaki Saic,
Ulrich Wiesnerc and
Ullrich Steiner§
*d
a,
Morgan Stefikb,
Hiroaki Saic,
Ulrich Wiesnerc and
Ullrich Steiner§
*d
aDepartment of Chemical Engineering, University College London, Torrington Place, London WC1E7JE, UK
bDepartment of Chemistry and Biochemistry, University of South Carolina, Columbia, South Carolina 29208, USA
cDepartment of Materials Science & Engineering, Cornell University, Ithaca, New York 14853, USA
dAdolphe Merkle Institute Switzerland, University of Fribourg, Chemin des Verdiers, 1700 Fribourg, Switzerland. E-mail: ullrich.steiner@unifr.ch
First published on 20th February 2015
Abstract
The coassembly of an inorganic sol by organic structure-directing agents is a widely established technique for solution processed mesoporous metal oxides. Block copolymers with highly amphiphilic blocks are promising candidates for fast and robust material assembly into a range of periodic nano-morphologies. The high polarity difference is however an inherent challenge for the simultaneous processing of hydrophilic and hydrophobic components. Here, we show that a solvent exchange through the evaporation of the initial volatiles and redissolution in an azeotrope mixture of polar and apolar solvents with a common high boiling point allows the evolution of a micellar solution towards block copolymer-dictated structures near thermodynamic equilibrium, with controlled access to inorganic cylinders, lamellae and organic cylinders within a continuous inorganic matrix.
Four requirements have to be fulfilled in order to reach equilibrium morphologies in blends of block copolymers with precursor sols during solvent evaporation: (I) the guest material has to exhibit selective interaction with one block of the polymer,1 (II) the inorganic material has to disperse into small units comparable to the size of the polymer chain,2 (III) the kinetics of the gelation process has to be slow compared to the duration of polymer phase separation,3 and (IV) a common solvent has to dissolve all components and evaporate sufficiently slowly to allow polymer self-assembly to equillibrate.4
While these conditions were soon met for the coassembly of silica-type materials,5,6 full structure control for metal oxide-based materials remains challenging. For TiO2, high chemical reactivity and multiple stable coordination states of the titanium precursor greatly limit the control over the kinetics of the sol–gel reaction. The fast hydrolysis of titanium alkoxide Ti(OR)4, by a factor of 105 compared to Si(OR)4, often impedes the block copolymer self-assembly.7 Routes to decelerate the gelation of the inorganic sol include the addition of stabilising ligands, low pH or non-hydrolytic synthesis.8 Usually an oversupply of aqueous HCl is added in order to maintain sol stability during block copolymer-directed material assembly.9 The coassembly of TiO2 by the widely studied block copolymer poly(ethylene oxide-block-propylene oxide-block-ethylene oxide) (Pluronic) is now well established in the bulk and in thin films,10,11 but most approaches suffer from long processing protocols and limited structure control. In principle, polymer blocks with a high interaction parameter χ are expected to be beneficial for fast and robust material synthesis by quickly driving self-assembly towards thermodynamic equilibrium. This should provide access to spherical, cylindrical, gyroid and lamellar morphologies in a range of metal oxide ceramics.
Alternatives to Pluronic, such as poly(ethylene-co-butylene-block-ethylene oxide) (KLE), poly(isoprene-block-ethylene oxide) (PI-b-PEO), poly(styrene-block-ethylene oxide) (PS-b-PEO) or poly(ethylene oxide-block-butadiene-block-ethylene oxide) (PEO-b-PB-b-PEO) have shown promising results in terms of larger pore sizes and temperature stability of the final material, but full structure-control for high TiO2 loadings is currently missing.12–18 Block copolymers with a large χ-value consist of strongly dissimilar chemical block, which poses a major challenge in terms of their solubility. Blending block copolymer solutions with excess water arising from the acid-stabilised TiO2 sol gives rise to microemulsions from which the volatile components evaporate with differing rates, leading to kinetically trapped morphologies that are far from thermodynamic equilibrium. A somewhat more elaborate route that circumvents this problem is a BCP-templating approach, where a BCP film is equilibrated and one of the blocks is removed to yield a mesoporous template that is subsequently backfilled with the inorganic material.19–21
We have recently introduced an additional step for the fabrication of PI-b-PEO-directed porous TiO2 films involving drying and redissolution of the block copolymer–sol mixture, which resulted in a greatly improved film quality and pore regularity.22,23 Here, we show that the redissolution of the as-made hybrid material in an azeotropic mixture of polar and apolar solvents with similar high boiling points allows the evolution from a micellar solution towards block copolymer-dictated structures near the thermodynamic equilibrium, with controlled access to inorganic cylinders, lamellae and hexagonal ceramic morphologies.
Our method is schematically shown in Fig. 1. A titanium-containing sol is prepared by the addition of 0.69 ml HCl (37%) to 1 ml titanium ethoxide (purum) under vigorous stirring.9 Depending on the inorganic to organic weight ratio, an appropriate amount of inorganic sol is subsequently mixed with 0.1 g PI-b-PEO block copolymer (molecular weight Mn = 33.5 kg mol−1, 23 wt% PEO (≈18.3 vol%), polydispersity index PDI = 1.03) dissolved in 7 ml of tetrahydrofuran (THF). For a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 between the resulting TiO2 and the amount of polymer in solution, 0.87 ml sol was added to 0.1 g BCP. The large amount of excess water arising from the acid-stabilisation of the metal oxide sol and high chemical dissimilarity of the hybrid components leads to a micellisation of the PI-block which is maintained during the evaporation of all volatile components. The dried material is subsequently redissolved in an azeotrope solvent mixture of 1-butanol (polar) and toluene (apolar) with a common high boiling point of TB = 105.6 °C.24 This solution is then dried under a glass dome at 40 °C. The reduction of excess water during the first evaporation step and the good solubility of all components in the high TB solvent mixture in the second cycle are essential for the slow evolution of microphase separation during solvent evaporation. Note that both evaporation cycles are required. Mixing of the TiO2 sol directly with an azeotrope solution of block copolymer leads to precipitation of the solute due to the high amount of water in the initial acid-stabilised hydrolytic sol.
:1 between the resulting TiO2 and the amount of polymer in solution, 0.87 ml sol was added to 0.1 g BCP. The large amount of excess water arising from the acid-stabilisation of the metal oxide sol and high chemical dissimilarity of the hybrid components leads to a micellisation of the PI-block which is maintained during the evaporation of all volatile components. The dried material is subsequently redissolved in an azeotrope solvent mixture of 1-butanol (polar) and toluene (apolar) with a common high boiling point of TB = 105.6 °C.24 This solution is then dried under a glass dome at 40 °C. The reduction of excess water during the first evaporation step and the good solubility of all components in the high TB solvent mixture in the second cycle are essential for the slow evolution of microphase separation during solvent evaporation. Note that both evaporation cycles are required. Mixing of the TiO2 sol directly with an azeotrope solution of block copolymer leads to precipitation of the solute due to the high amount of water in the initial acid-stabilised hydrolytic sol.
 | ||
| Fig. 1 Schematic of the fabrication route. (a) An acid-stabilised hydrolytic TiO2 sol and a PI-b-PEO block copolymer are mixed in THF. (b) The block copolymer forms micelles with PI cores that are preserved during evaporation of all volatile components. (c) Redissolution of this material in an azeotrope mixture of polar and apolar solvents with a common high boiling point overcomes micellisation and enables the evolution of the block copolymer–sol blend towards thermodynamic equilibrium. | ||
The resulting morphology of a self-assembled hybrid material is shown in the transmission electron microscopy (TEM) images in Fig. 2. All bulk samples were cross-sectioned by cryo-ultramicrotomy to allow TEM imaging. The one-stage evaporation procedure, where a mixed solution was slowly dried in a Petri dish at 40 °C under a glass dome led to a micellar arrangement as shown in Fig. 2a. This morphology is similar to those reported for the KLE system.25 The effect of a two-stage process, with evaporation of primary volatiles and azeotrope redissolution, is shown in Fig. 2b. An inverse hexagonal morphology with a matrix of PEO/TiO2 containing PI cylinders was identified. Different projections of the cylinders are visible in the image plane, clearly distinguishing this morphology from the micellar array of Fig. 2a. The average cylinder-to-cylinder spacing determined by electron microscopy was 28 ± 3 nm. This observation is supported by small-angle X-ray scattering (SAXS) data, shown in Fig. 2c. While the radially integrated scattering pattern of the one-stage evaporated sample is consistent with a close-packed micellar arrangement of pores (exhibiting an appreciable degree of polydispersity), the reflection peaks of the redissolved sample are clearly shifted and consistent with the allowed positions for an arrangement of hexagonally packed cylinders. A characteristic length scale of d = 2π/q = 52 nm can be derived from scattering pattern of the micellar array while the SAXS data after solvent exchange corresponds to a cylinder-to-cylinder spacing of 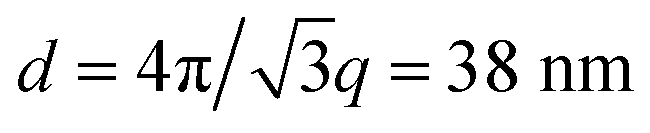 .26 This is a convincing indication that the evaporation of primary volatiles and redissolution resulted in improved solubility of the polymer–sol blend, which in combination with greatly decelerated evaporation kinetics led to a microphase separated hybrid material in close resemblance with the block copolymer morphology near thermodynamic equilibrium.
.26 This is a convincing indication that the evaporation of primary volatiles and redissolution resulted in improved solubility of the polymer–sol blend, which in combination with greatly decelerated evaporation kinetics led to a microphase separated hybrid material in close resemblance with the block copolymer morphology near thermodynamic equilibrium.
 | ||
| Fig. 2 Effect of redissolution on bulk coassembly of PI-b-PEO & TiO2 from a precursor sol. TEM images of samples (a) without and (b) with the described redissolving step under otherwise identical experimental conditions. The scale bars represent 200 nm. (c) Radially integrated SAXS patterns of both samples. The vertical lines correspond to the expected peak positions for a micellar array (black) and hexagonally packed cylinders (red). | ||
The versatility of this approach is demonstrated in Fig. 3, where the mixing ratio of the block copolymer to titania-precursor in the initial solution was varied. The TEM images reveal that a 1:3 ratio (defined by the mass of the resulting TiO2 after calcination to the mass of polymer in solution) led to the formation of TiO2-rich cylinders surrounded by a PEO matrix, with a characteristic spacing of 44 ± 7 nm. The vol% of the PI block used in this study was 82%, indicating that the incorporated TiO2 must have swollen the PEO-rich phase to drive the hydrophilic block into a hexagonal arrangement of cylinders.27 The observed morphology is consistent with SAXS data in Fig. 3c, having a cylinder-to-cylinder spacing of 36 nm. The predicted reflection peak positions for a hexagonal morphology are indicated with black lines. Increasing the inorganic weight fraction to 1:2 resulted in a morphology resembling a lamellar arrangement of the components with a characteristic spacing of 43 ± 2 nm, shown in Fig. 3b. This is in line with SAXS where the observed peaks are consistent with allowed reflections for a lamellar phase having a layer spacing of 36 nm. A further increase of the TiO2 weight fraction to 2:1 led to an inverse hexagonal morphology as discussed previously (Fig. 2). In fact, the presented two-step procedure enabled high inorganic loading up to mass ratios of 5:1.
 | ||
| Fig. 3 Self-assembly in the hybrid composite. (a and b) TEM images of PI-b-PEO/TiO2 samples. The specimen were cast from inorganic to organic mixing ratios of (a) 1:3 and (b) 1:2, which led to two different morphological arrangements, TiO2-rich cylinders in an organic matrix and TiO2 lamellae, respectively. The scale bars represent 500 nm. (c and d) Corresponding radially integrated SAXS spectra. The expected peak positions for (c) a hexagonal arrangement of cylinders and (d) lamellae are indicated as black lines, calculated from the position of the first reflection peak. | ||
We observed a 17–19% difference between the characteristic spacings determined by TEM analysis compared to SAXS analysis. Where values of 36 nm, 36 nm and 38 nm were determined for the 1:3, 1:2 and 2:1 ratios by SAXS, analysis of the TEM images led to values of 44 ± 7, 43 ± 2 nm, and 28 ± 3 nm, respectively (see ESI†). The reasons for these deviations include off-axis imaging of the lamellae, stretching the apparent spacing observed by TEM. Symmetry considerations exclude this cause for the measured regions of the hexagonal and inverse hexagonal morphologies. Our observations are consistent with earlier reports specifying microtome-induced distortion and PI reflow after microtoming as likely causes of these differences.6,28
The results presented in Fig. 2 and 3 illustrate the potential of introducing a redissolution step after partial evaporation of the primary volatiles. The success of this approach depends, however, on the appropriate timing for evaporation and redissolution. If the condensation reaction arrests the material during the first evaporation step, it will no longer redissolve. On the other hand, interrupting the primary evaporation process too early may cause a deterioration of the structure formation process by residual water. In experimental practise, the appropriate timing proved relatively robust. The primary evaporation process was stopped when a sudden increase in viscosity inhibited further flow of residual solvent in the Petri dish.
In an earlier publication by Garcia et al. the effects of the block copolymer composition as well as the inorganic to organic ratio were discussed in detail for a PI-b-PEO aluminosilicate sol mixture, where the kinetics of the sol–gel chemistry are somewhat less challenging.29 A ternary phase diagram with resulting equilibrium morphologies as a function of the volume ratio of PI, PEO and inorganic content was presented. Consistent with this previous publication, we have observed that gradually changing the inorganic to organic weight ratio from 1:4 to 9:1 led to changes of the resulting sample morphologies, from inorganic cylinders, to lamellae, and finally an inverse hexagonal morphology. We anticipate that the presented approach of redissolution will enable the generation of a similar phase diagram for PI-b-PEO/TiO2.
In summary, we present a powerful method to compatibilise the coassembly of a non-water soluble block copolymer with hydrolytic sol–gel chemistry. The introduction of a solvent exchange step enabled the surmounting of kinetic barriers from trapped solution phases. As a result, hybrid material arrangement of hydrophobic PI and hydrophilic PEO/TiO2 closely resemble the corresponding block copolymer melt morphologies at thermodynamic equilibrium. The variation of the inorganic guest and the polymer host mixing ratio in solution enabled access to a range of morphologies including hexagonal TiO2-rich cylinders in an organic matrix, TiO2 lamellae and a TiO2 inverse hexagonal matrix.
Acknowledgements
This publication is based on work supported by the National Science Foundation (DMR-1409105). SG is grateful for support by the German National Academy of Sciences Leopoldina, Fellowship LPDS2012-13. MS acknowledges startup funds from the University of South Carolina.References
- P. Simon, R. Ulrich, H. Spiess and U. Wiesner, Chem. Mater., 2001, 13, 3464–3486 CrossRef CAS.
- S. Warren, F. Disalvo and U. Wiesner, Nat. Mater., 2007, 6, 156–161 CrossRef CAS PubMed.
- J. Fan, S. W. Boettcher and G. D. Stucky, Chem. Mater., 2006, 18, 6391–6396 CrossRef CAS.
- D. Grosso, F. Cagnol, G. Soler-Illia, E. Crepaldi, H. Amenitsch, A. Brunet-Bruneau, A. Bourgeois and C. Sanchez, Adv. Funct. Mater., 2004, 14, 309–322 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. Fredrickson, B. Chmelka and G. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- M. Templin, A. Franck, A. DuChesne, H. Leist, Y. Zhang, R. Ulrich, V. Schädler and U. Wiesner, Science, 1997, 278, 1795–1798 CrossRef CAS.
- C. Brinker and G. Scherer, Sol–Gel Science: The Physics and Chemistry of Sol–Gel Processing, Academic Press, 1st edn, 1990 Search PubMed.
- S. Boettcher, J. Fan, C.-K. Tsung, Q. Shi and G. Stucky, Acc. Chem. Res., 2007, 40, 784–792 CrossRef CAS PubMed.
- P. Alberius, K. Frindell, R. Hayward, E. Kramer, G. Stucky and B. Chmelka, Chem. Mater., 2002, 14, 3284–3294 CrossRef CAS.
- E. Crepaldi, G. Soler-Illia, D. Grosso, F. Cagnol, F. Ribot and C. Sanchez, J. Am. Chem. Soc., 2003, 125, 9770–9786 CrossRef CAS PubMed.
- C. Sanchez, C. Boissière, D. Grosso, C. Laberty and L. Nicole, Chem. Mater., 2008, 20, 682–737 CrossRef CAS.
- B. Smarsly, D. Grosso, T. Brezesinski, N. Pinna, C. Boissière, M. Antonietti and C. Sanchez, Chem. Mater., 2004, 16, 2948–2952 CrossRef CAS.
- J. Lee, M. Orilall, S. Warren, M. Kamperman, F. DiSalvo and U. Wiesner, Nat. Mater., 2008, 7, 222–228 CrossRef CAS PubMed.
- J. Zhang, Y. Deng, D. Gu, S. Wang, L. She, R. Che, Z.-S. Wang, B. Tu, S. Xie and D. Zhao, Adv. Energy Mater., 2011, 1, 241–248 CrossRef CAS.
- E. Ortel, A. Fischer, L. Chuenchom, J. Polte, F. Emmerling, B. Smarsly and R. Kraehnert, Small, 2012, 8, 298–309 CrossRef CAS PubMed.
- Y. Deng, J. Wei, Z. Sun and D. Zhao, Chem. Soc. Rev., 2013, 42, 4054–4070 RSC.
- J. Hwang, C. Jo, K. Hur, J. Lim, S. Kim and J. Lee, J. Am. Chem. Soc., 2014, 136, 16066–16072 CrossRef CAS PubMed.
- D. Fattakhova-Rohlfing, A. Zaleska and T. Bein, Chem. Rev., 2014, 114, 9487–9558 CrossRef CAS PubMed.
- B. W. Boudouris, C. D. Frisbie and M. A. Hillmyer, Macromolecules, 2008, 41, 67–75 CrossRef CAS.
- K. Sivanandan, T. Chatterjee, N. Treat, E. J. Kramer and C. J. Hawker, Macromolecules, 2010, 43, 233–241 CrossRef CAS.
- S. Vignolini, N. A. Yufa, P. S. Cunha, S. Guldin, I. Rushkin, M. Stefik, K. Hur, U. Wiesner, J. J. Baumberg and U. Steiner, Adv. Mater., 2012, 24, OP23–OP27 CrossRef CAS PubMed.
- S. Guldin, S. Hüttner, P. Tiwana, M. Orilall, B. Ülgüt, M. Stefik, P. Docampo, M. Kolle, G. Divitini, C. Ducati, S. Redfern, H. Snaith, U. Wiesner, D. Eder and U. Steiner, Energy Environ. Sci., 2011, 4, 225–233 CAS.
- S. Guldin, M. Kolle, M. Stefik, R. Langford, D. Eder, U. Wiesner and U. Steiner, Adv. Mater., 2011, 23, 3664–3668 CrossRef CAS PubMed.
- J. Ponton, Azeotrope Databank, http://eweb.chemeng.ed.ac.uk/, 2001.
- A. Thomas, H. Schlaad, B. Smarsly and M. Antonietti, Langmuir, 2003, 19, 4455–4459 CrossRef CAS.
- S. Foerster, S. Fischer, K. Zielske, C. Schellbach, M. Sztucki, P. Lindner and J. Perlich, Adv. Colloid Interface Sci., 2011, 163, 53–83 CrossRef CAS PubMed.
- C. Park, J. Yoon and E. Thomas, Polymer, 2003, 44, 6725–6760 CrossRef CAS PubMed.
- M. Stefik, S. Wang, R. Hovden, H. Sai, M. W. Tate, D. A. Muller, U. Steiner, S. M. Gruner and U. Wiesner, J. Mater. Chem., 2012, 22, 1078–1087 RSC.
- B. Garcia, M. Kamperman, R. Ulrich, A. Jain, S. Gruner and U. Wiesner, Chem. Mater., 2009, 21, 5397–5405 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra00836k |
| ‡ Present address: Simpson Querrey Institute for BioNanotechnology (SQI), Northwestern University, Evanston, IL 60208, USA |
| § Present address: Adolphe Merkle Institute, University of Fribourg, Chemin des Verdiers, 1700 Fribourg, CH. Email: E-mail: ullrich.steiner@unifr.ch |
| This journal is © The Royal Society of Chemistry 2015 |