
Biodegradable multi-liposomal containers†
Alexander A. Yaroslavov*a,
Anna A. Efimovaa,
Andrey V. Sybachina,
Sergey N. Chvalunb,
Alevtina I. Kulebyakinab and
Ekaterina V. Kozlovab
aLomonosov Moscow State University, 119991, Leninskie Gory, 1-3, Moscow, Russia. E-mail: yaroslav@genebee.msu.ru; Fax: +74959390174; Tel: +74959393116
bNational Research Centre “Kurchatov Institute”, 123182, Akademika Kurchatova sq., 1, Moscow, Russia. E-mail: presscentr@kiae.ru; Fax: +74991967038; Tel: +74991967038
First published on 18th March 2015
Abstract
Here we describe biodegradable multi-liposomal core–shell containers. They are made of a poly-L-lactide–polyethyleneoxyde–poly-L-lactide triblock-copolymer core covered by electrostatically adsorbed liposomes. The liposomes retain their integrity after adsorption, which allows them to be used for encapsulating bioactive compounds. The multi-liposomal containers eventually decompose, by being attacked by hydrolytic enzymes, which makes them promising for use in drug delivery.
Introduction
Bilayer lipid vesicles (liposomes) are currently used for delivery of biologically active substances: in particular drugs,1–3 polypeptides and proteins,4–6 and nucleic acids.7–9 Multi-liposome assembly, for example via immobilization of several liposomes on a nano-sized colloid, can increase the efficacy of liposome uptake by cells and the therapeutic effect of a liposomal drug. However, attachment of liposomes to a solid carrier (glass, ceramic, metal, polymer, etc.) is usually accompanied by their destruction and the uncontrolled release of the encapsulated drugs.10 A few successful examples of intact liposome immobilization described in the literature include pre-modification of both liposomes and the surface of a solid carrier.11 There nevertheless remains a need for the development of carriers that do not have the above-mentioned disadvantages.Recently, we have proposed to adsorb anionic liposomes on the surface of polystyrene microspheres with grafted polycationic chains, i.e., “spherical polycationic brushes”.12 It was shown that a mixture of liposomes with different contents could be immobilized on the brush surface.13 This method allows dozens of liposomes to be concentrated within a rather small volume.14 The hydrophilic layer of grafted macromolecules ensures the integrity of the immobilized liposomes by preventing them from directly contacting the solid polystyrene.12 Multi-liposomal containers (MLCs) prepared in this manner can be used for constructing working in vitro catalysts, fluorescent markers, and analytical systems. Their in vivo application, however, is strongly restricted by the non-biodegradability of the polystyrene core of the brushes.
In the present work we describe MLCs, approx. 400 nm in diameter, based on biodegradable polylactide (PLA) particles stabilized by polyethyleneglycol (PEG) chains.15 Here, liposomes are adsorbed electrostatically on the PLA particle surface. Polylactide is destroyed in the presence of hydrolytic enzymes (esterases),16,17 rendering the entire MLC biodegradable. MLCs made in this manner look to be promising for “passive targeting” due to selective penetration of particles with dimensions of 200–400 nm in the capillaries of tumors and other inflamed areas.18
Experimental
Materials
Cardiolipin (CL2−) and egg lecithin (EL) from Avanti and polylysine hydrobromide with a degree of polymerization equal to 340 from Sigma were used as received. Poly(N-ethyl-4-vinylpyridinium bromide) (PEVP) was synthesized via quaternization of poly(4-vinylpyridine) with a degree of polymerization equal to 600 (Sigma) by ethyl bromide as described elsewhere.19 The resulting product was actually a copolymer containing 95 mol% of quaternized pyridinium and 5 mol% of residual 4-vinylpyridine units as determined according to IR spectroscopy.19 Concentrations of cationic polymers are given in moles of amino-groups (for polylysine) and quaternized units (for PEVP) per liter.To hydrolyze the ester bonds in PLA, the proteolytic complex morikrase from the hepatopancreas of the Kamchatka crab Paralithodes camchatica (JSC RPE Trinita, Russia) was used.20,21 Morikrase is actually a mixture of enzymes including serine proteinases, collagenases, metalloproteinases, etc., capable of cleaving ester and peptide/amide bonds. Morikrase shows enzymatic activity in a pH region from 6.0 to 9.0 with optimum activity at pH 7.5.
Triblock-copolymer of poly-L-lactide and poly(ethylene glycol) (Fig. 1) was synthesized via ring-opening polymerization of lactide in the presence of PEG, catalyzed by stannous octanoate.22 The structure and purity of the copolymer was confirmed by NMR, gel chromatography and IR spectroscopy (see details in ESI†). The molecular mass of the copolymer was found to be 11.380 Da with a polydispersity index equal to 1.4. To prepare an aqueous solution of copolymer, a standard procedure was applied.23 Briefly, the copolymer was dissolved in tetrahydrofuran to attain a copolymer concentration of 5 × 10−5 M. Distilled water was added the next day under intensive stirring upto 10 vol% water content. After another day, distilled water was again added under intensive stirring up to 20 vol% water content. The water–tetrahydrofuran mixture prepared in this manner was dialyzed against distilled water for one week to remove the organic solvent. Copolymer concentration is given in moles of copolymer macromolecule per liter.
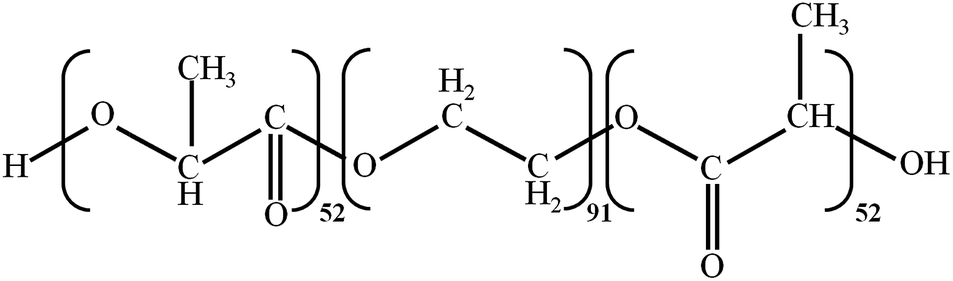 | ||
| Fig. 1 Triblock-copolymer of poly-L-lactide and poly(ethylene glycol). | ||
Small unilamellar anionic liposomes, 50–60 nm in diameter, were prepared by the standard sonication procedure.24 Briefly, a mixed EL/CL2− chloroform solution was evaporated under vacuum, and the resulting thin lipid film was dispersed in a Tris buffer (pH 7, 10−2 M) for 400 s with a 4700 Cole-Parmer ultrasonic homogenizer. Liposome samples were separated from titanium dust by centrifugation for 5 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, and used within one day. Mixed EL/CL2− liposomes with a molar fraction of anionic CL2− head-groups ν = 2[CL2−]/(2[CL2−] + [EL]) = 0.1 were thus obtained.
000 rpm, and used within one day. Mixed EL/CL2− liposomes with a molar fraction of anionic CL2− head-groups ν = 2[CL2−]/(2[CL2−] + [EL]) = 0.1 were thus obtained.
To prepare liposomes loaded by a NaCl solution, we followed the procedure described in ref. 25. The lipid film was suspended in a mixed 1 M NaCl/10−3 M Tris buffer solution and extruded through polycarbonate membranes with an average pore diameter of 50 nm, using Avanti's mini-extruder. Then the liposome suspension was separated from excess NaCl by dialysis for 4.5 hours, with the outer 10−3 M Tris buffer solution changed every 1.5 hours. The integrity of the NaCl-loaded liposomes was assessed conductometrically as described in ref. 12.
Double-distilled water was used for making solutions after additionally treating it with a Milli-Q Millipore system composed of ion-exchange and adsorption columns as well as a filter to remove large particles. All experiments were carried out at 20 °C.
Methods
Mean hydrodynamic diameters of particles were determined by dynamic light scattering at a fixed scattering angle of 90° in a thermostatic cell with a Brookhaven Zeta Plus instrument. Software provided by the manufacturer was employed to calculate diameter values. In each experiment, fresh liposomes were used, whose size (hydrodynamic diameter) fluctuated from sample to sample but always fell within the 40–60 nm range with a mean value of 50 nm. The electrophoretic mobility (EPM) of the particles was measured by laser microelectrophoresis in a thermostatic cell using a Brookhaven Zeta Plus instrument with the corresponding software.The decomposition of the PLA–polylysine–liposome ternary complex was initiated by addition of 0.05 mg ml−1 of the proteolytic complex morikrase. Then the particle size was measured by means of dynamic light scattering.
The pH values of solutions were measured using a pH meter 210 (Hanna, Germany); and the conductivities of the solutions were measured using a CDM 83 conductometer (Radiometer, Denmark).
Results and discussion
Biodegradable PLA particles were used for constructing the MLC. In a 10−2 M aqueous Tris buffer solution, the average hydrodynamic diameter (size) and EPM of the particles was found to be equal to 160 nm and −0.6 (μm s−1)/(V cm−1), respectively. A small negative charge was probably imparted due to dissociation of terminal OH-groups of the copolymer.Liposomal containers were prepared from an EL/CL2− mixture with a molar fraction of anionic CL2− head-groups ν = 0.1 as described in the Experimental section. To make anionic EL/CL2− liposomes capable of binding to the negative PLA particles, they were modified by a cationic polymer, PEVP or polylysine. Fig. 2 (curve 1) shows the dependence of the EPM of EL/CL2− liposomes on the concentration of cationic PEVP in the system. Addition of PEVP was accompanied by neutralization of the liposome surface charge and decrease of the EPM down to zero, then the liposome surface became positive, and finally the EPM reached a positive charge at a [PEVP] = 2.5 × 10−4 M.
 | ||
| Fig. 2 Electrophoretic mobility (EPM) (1) and hydrodynamic diameter (2) of PEVP–EL/CL2− liposome complexes vs. PEVP concentration. νCL = 0.1; total lipid concentration, 1 mg ml−1; Tris buffer, 10−2 M; pH 7.0. Point Ⓐ corresponds to the PEVP concentration in MOCO used for complexation with PLA particles. | ||
In parallel, the average size of EL/CL2− liposomes in the presence of PEVP was measured using dynamic light scattering (Fig. 2, curve 2). The liposome size was observed to increase with an increase in PEVP concentration, reaching a maximum at an EPM = 0, and decreasing at higher PEVP concentrations. Both the EPM and light scattering data show that electrostatic adsorption of positive PEVP occurred on the surface of negative liposomes. These results are in agreement with earlier published data on aggregation of liposomes triggered by cationic polymers.19,26,27
In order to estimate the efficacy of the binding of PEVP to liposomes, the liposomes with adsorbed PEVP were separated by centrifugation and the supernatants were analyzed spectrophotometrically (see details in ESI†). The dependence of the PEVP concentration in the supernatant on the initial PEVP concentration (Fig. 3) indicates a complete binding of PEVP to liposomes up to a [PEVP] = 2.5 × 10−4 M. This point corresponds to the maximum positive charge of the PLA particles (Fig. 2, curve 1) that leads to an electrostatic barrier on the PLA particle surface and termination of further PEVP adsorption.
 | ||
| Fig. 3 PEVP concentration in the supernatant after centrifugation of the PEVP–EL/CL2− liposome complexes vs. initial PEVP concentration. νCL = 0.1; total lipid concentration, 1 mg ml−1; Tris buffer, 10−2 M; pH 7.0. Point Ⓐ corresponds to the PEVP concentration in MOCO used for complexation with PLA particles. | ||
For complexation with PLA particles, a PEVP–liposome complex was prepared by mixing a solution of 1.7 × 10−4 M PEVP with a 1 mg ml−1 EL/CL2− liposome suspension. This complex was positively charged (point A on curve 1, Fig. 2) and all added PEVP was involved in complexation with PLA particles (point A in Fig. 3). Modifying complex (MOCO) prepared in this manner was determined to contain four PEVP chains per liposome. The mean size of the complex particles was measured by dynamic light scattering to be 170 nm.
The integrity of liposomes in MOCO was controlled conductometrically. Suspensions of liposomes loaded with a 1 M NaCl solution were prepared. A release of NaCl from liposomes into surrounding solution was accompanied by an increase in the conductivity (Ω) of the suspension. The results were compared with the conductivity of a suspension of NaCl-loaded liposomes completely destroyed in the presence of excess surfactant (Triton X-100); this latter level of conductivity was defined as 100% activity (Ωmax). It was found that the conductivity did not change (i.e., the liposome integrity was retained) up to 3 hours after MOCO formation.
MOCO was then added to a suspension of the PLA particles. Its binding to PLA was controlled by measuring the average size of the particles in the system. The results are given in Fig. 4 (curve 1): on the X-axis is shown the molar concentration of PEVP in MOCO added to the PLA particles ([PEVP]MOCO). An increase in PEVP concentration resulted in an increase of particle size that reflected formation of a PLA–PEVP–liposome ternary complex. The maximum particle size was reached at a [PEVP]MOCO = 6.8 × 10−5 M.
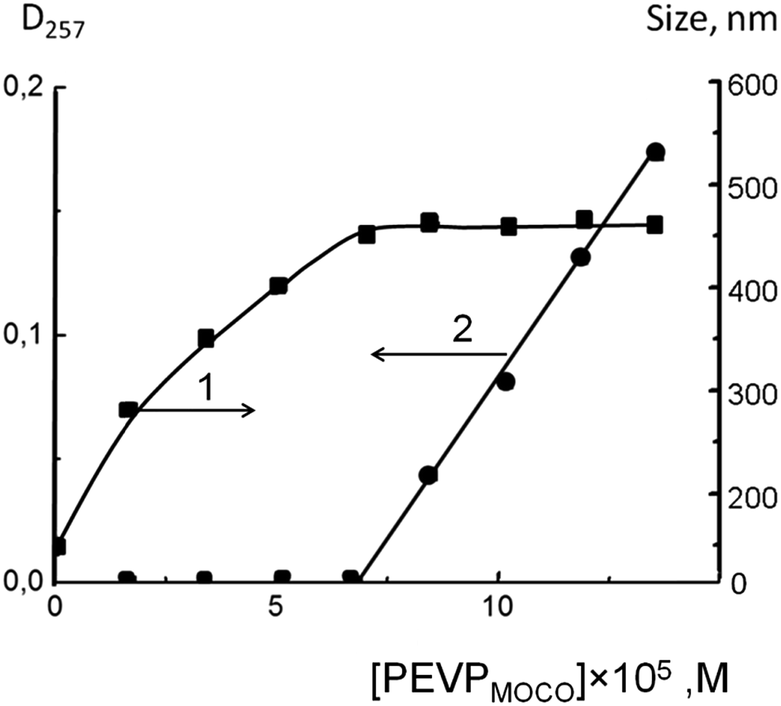 | ||
| Fig. 4 Hydrodynamic diameter (1) and optical density at 257 nm (2) of PLA–PEVP–liposome ternary particles vs. PEVP concentration in MOCO. EL/CL2− liposomes, νCL = 0.1; Tris buffer, 10−2 M; pH 7.0. Ccopolymer = 10−5 M. | ||
Additionally, the binding of MOCO to the PLA particles was analyzed spectrophotometrically as shown above for the PEVP–liposome binary system. After separation of the ternary complex, optical densities in the supernatants at 257 nm were measured, and observed to correspond to the maximum adsorption in the UV spectrum of PEVP. At the same wavelength, we had to take into account the scattering of liposomes (or their complexes with PEVP) not bound to the PLA particles. According to the plot of optical density vs. [PEVP]MOCO presented in Fig. 4 (curve 2), there is a rather wide range of polycation concentrations ([PEVP]MOCO ≤ 6.8 × 10−5 M) with zero optical density, indicating that all added MOCO was bound to the PLA particles up to this concentration. At this PEVP concentration, PLA–PEVP–liposome ternary particles reached their maximum size (curve 1 in Fig. 4).
Additionally, the binding of MOCO to PLA was followed by using fluorescence. PEVP is known to be a strong fluorescence quencher.28 The binding of PEVP to EL/CL2− liposomes with the fluorescent label (N-fluorescein-iso-thiocyanyldipalmitoyl-phosphatidylethanolamine) incorporated into the lipid bilayer was accompanied by a decrease of the fluorescence. When MOCO with a quenched fluorescence was added to a micellar PLA solution, no change in the fluorescence intensity was detected, indicating the binding of non-dissociated MOCO to the PLA particles.
The integrity of liposomes in the ternary complex was evaluated using an earlier described conductometry method. MOCO with a [PEVPMOCO] = 3.4 × 10−5 M and 0.2 mg ml−1 liposome was added to the PLA suspension. The conductivity was monitored for up to 3 hours after mixing the components. It was found that the conductivity of the suspensions of the ternary complexes did not change in this time period, which means that the integrity of the liposomes involved in the ternary complex formation remained unchanged.
The composition of the ternary complex can be rationalized as follows. According to ref. 22, 29 and 30, PLA particles dissolved in water are micellar-like structures with a core composed of PLA blocks partly crystallized into unstable micro-crystallites and a PEG shell exposed to water, thus ensuring the aggregation stability of the micelles in the surrounding water. Each PLA block is assumed to form an α-crystal (a = 1.07 nm, b = 0.615 nm, c = 2.78 nm) consisting of 10/3 helical chains.31 We examined the PLA–PEG copolymer with the use of calorimetry and atomic force microscopy (AFM). Using calorimetry, a peak corresponding to melting at 145 °C was revealed for the copolymer that coincided with the melting point for homo-PLA polymer found earlier, which confirmed a crystalline structure of the PLA core in the PLA–PEG copolymer.29 AFM showed the thickness of the PLA–PEG particles (micelles) immobilized on the mica surface to be 10–15 nm. Bases on these data, two possible packing arrangements of the PLA chains can be proposed: with the chains either extended, which would yield a thickness of 14 nm, or double folded, yielding a thickness of 7 nm (Fig. 5). In the case of the more folded chains, the size of the primary nucleus would be too small. Thus, the area of one copolymer macromolecule with an extended conformation in the micelle would be 1.07 nm × 0.615 nm × 2 ≈ 1.3 nm2, while that with a double folded conformation would occupy an area of 2.6 nm2. The total area of the copolymer micelles with extended chains can be calculated as Sm = 1.3 nm2 × Ccopolymer × A = 0.8 × 1019 nm2 and with double folded chains as Sm = 1.6 × 1019 nm2, where Ccopolymer = 10−5 M, and A is Avogadro's number.
 | ||
| Fig. 5 A chain-packing model of the PLA crystals with extended (a) and double folded (b) chains. | ||
Saturation of the copolymer micelles with MOCO was achieved at a [PEVP]MOCO = 6.8 × 10−5 M (Fig. 4, curve 2), which corresponds to the concentration of liposomes in MOCO equal to 0.4 g L−1 or 1.3 × 1016 liposomes per liter. Assuming an area covered by each adsorbed liposome equal to 3.14 × 25 nm × 25 nm = 1.9 × 103 nm2, the total area that can be occupied by adsorbed liposomes Slip = 1.9 × 103 nm2 × 1.3 × 1016 = 2.6 × 1019 nm2.
We see therefore a good correlation between the copolymer micelle area available for liposome binding (Sm) and the area covered by liposomes at saturation (Slip): Sm ≈ Slip. Actually, Slip is less than Sm because of adsorption of liposome–PEVP (MOCO) aggregates but not of individual liposomes.
The PLA–PEVP–liposome ternary complex described above contains a biodegradable core made up of PLA particles and a shell of electrostatically bound to PLA peripheral liposomes. However, the third “intermediate” component, PEVP, does not decompose down to smaller fragments in a biological environment. In order to get around this difficulty, we replaced PEVP with a biodegradable cationic polymer, polylysine. This polycation was found to quantitatively complex anionic EL/CL2− liposomes similar to PEVP. In excess polylysine, the complex acquired a positive charge and became adsorbed on the surface of the anionic PLA particles. Fig. 6 shows how the size of the particles in the system changed when the polylysine–liposome modifying complex (MOCO), with 4 polylysine chains per liposome, bound to the PLA particles. An increase of polylysine concentration resulted in an increase of particle size, with the maximum particle size reached at a polylysine concentration in the binary complex, [polylysine]MOCO = 4.3 × 10−5 M. As indicated by the conductometrical experiments with NaCl-loaded liposomes, their integrity remained unchanged after binding to polylysine and subsequent adsorption of the polylysine–liposome MOCO on the surfaces of the PLA particles.
 | ||
| Fig. 6 Hydrodynamic diameter of PLA–polylysine–liposome ternary particles vs. polylysine concentration in MOCO. EL/CL2− liposomes, νCL = 0.1; Tris buffer, 10−2 M; pH 7.0. Ccopolymer = 10−5 M. | ||
All components of the PLA–polylysine–liposome ternary complex are biodegradable, which allows for its decomposition in a biological environment. The decomposition was initiated by addition of the proteolytic complex morikrase, which is capable of cleaving ester bonds in PLA and lipid molecules and amide bonds in polylysine, to a suspension of the ternary complex. This decomposition was measured using dynamic light scattering. The hydrolytic process was evaluated by measuring the average size of the particles in the suspension. In a control experiment without morikrase, no changes in the average size of the ternary complex particles were detected for up to a week after preparation of the complex. In contrast, addition of morikrase to the ternary complex initiated a decrease in its size down to 10–15 nm within 100 hours (4 days) (Fig. 7). This size is much less than that observed for either initial component: 160 nm for PLA, 200 nm for the polylysine/liposome binary complex, and 60 nm for the liposomes. This result definitively shows enzyme-induced biodegradation of the PLA–liposome complex.
 | ||
| Fig. 7 The kinetics of the decrease in the average hydrodynamic diameter of PLA–polylysine–liposome ternary particles after addition of morikrase. [Polylysine]MOCO = 4.3 × 10−5 M. [Morikrase] = 0.05 mg ml−1; EL/CL2− liposomes, νCL = 0.1; Tris buffer, 10−2 M; pH 7.0. Ccopolymer = 10−5 M. | ||
Biodegradable multi-liposomal containers composed of a PLA core and electrostatically adsorbed liposomes have been described. The liposomes retain their integrity after adsorption, which allows them to be used to encapsulate bioactive compounds. The multi-liposomal containers eventually decompose after being attacked by hydrolytic enzymes, which makes them promising for use in the field of drug delivery.
Acknowledgements
This work was supported by Russian Science Foundation (project 14-13-00255). The authors are grateful to Professor Galina N. Rudenskaya (Lomonosov Moscow State University, Russia) for the morikrase sample and for stimulating discussions.Notes and references
- M. L. Immordino, Int. J. Nanomed., 2006, 1, 297–315 CrossRef CAS PubMed.
- A. Madni, M. Sarfraz, M. Rehman, M. Ahmad, N. Akhtar, S. Ahmad, N. Tahir, S. Ijaz, R. Al-Kassas and R. J. Löbenberg, J. Pharm. Pharm. Sci., 2014, 17, 401–426 CAS.
- V. Torchilin and V. Weissig, Liposomes: A Practical Approach, Oxford Univ. Press, Oxford, 2nd edn, 2003 Search PubMed.
- M. S. Kim, D. W. Lee, K. Park, S. J. Park, E. J. Choi, E. S. Park and H. R. Kim, Colloids Surf., B, 2014, 116, 17–25 CrossRef CAS PubMed.
- L. E. Santos, M. C. Colhone, K. R. Daghastanli, R. G. Stabeli, I. Silva-Jardim and P. Ciancaglini, J. Colloid Interface Sci., 2009, 340, 112–118 CrossRef CAS PubMed.
- F. A. do Carmo, P. C. Sathler, P. Zancan, C. R. Rodrigues, H. C. Castro, V. P. de Sousa, M. Sola-Penna and L. M. Cabral, Curr. Pharm. Biotechnol., 2014, 15, 620–628 CAS.
- C. M. Knapp and K. A. Whitehead, Expert Opin. Drug Delivery, 2014, 11, 1923–1937 CrossRef CAS PubMed.
- Y. Yamada, M. Tabata, Y. Yasuzaki, M. Nomura, A. Shibata, Y. Ibayashi, Y. Taniguchi, S. Sasaki and H. Harashima, Biomaterials, 2014, 35, 6430–6438 CrossRef CAS PubMed.
- A. Ewe, A. Schaper, S. Barnert, R. Schubert, A. Temme, U. Bakowsky and A. Aigner, Acta Biomater., 2014, 10, 2663–2673 CrossRef CAS PubMed.
- R. P. Richter, R. Berat and A. R. Brisson, Langmuir, 2006, 22, 3497–3505 CrossRef CAS PubMed.
- H. Chen, Y. Teramura and H. Iwata, Biomaterials, 2011, 32, 7971–7977 CrossRef CAS PubMed.
- A. A. Yaroslavov, A. V. Sybachin, M. Schrinner, M. Ballauff, L. Tsarkova, E. Kesselman, J. Schmidt, Y. Talmon and F. M. Menger, J. Am. Chem. Soc., 2010, 132, 5948–5949 CrossRef CAS PubMed.
- A. V. Sybachin, O. V. Zaborova, M. Ballauff, E. Kesselman, J. Schmidt, Y. Talmon, F. M. Menger and A. A. Yaroslavov, Langmuir, 2012, 28, 16108–16114 CrossRef CAS PubMed.
- A. V. Sybachin, O. V. Zaborova, V. N. Orlov, P. I. Semenyuk, M. Ballauff, E. Kesselman, J. Schmidt, Y. Talmon, F. M. Menger and A. A. Yaroslavov, Langmuir, 2014, 30, 2441–2447 CrossRef CAS PubMed.
- T. Fujiwara, M. Miyamoto, Y. Kimura, T. Iwata and Y. Doi, Macromolecules, 2001, 34, 4043–4050 CrossRef CAS.
- K. Madhavan Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef CAS PubMed.
- S. M. Reeve, S. P. McCarthy, M. J. Downey and R. A. Gross, Macromolecules, 1994, 27, 825–831 CrossRef.
- A. Laouini, C. Jaafar-Maalej, I. Limayem-Blouza, S. Sfar, C. Charcosset and H. J. Fessi, J. Colloid Sci. Biotechnol., 2012, 1, 147–168 CrossRef PubMed.
- A. A. Yaroslavov, T. A. Sitnikova, A. A. Rakhnyanskaya, Yu. A. Ermakov, T. V. Burova, V. Ya. Grinberg and F. M. Menger, Langmuir, 2007, 23, 7539–7544 CrossRef CAS PubMed.
- G. N. Rudenskaya, Bioorg. Khim., 2003, 29, 117–128 Search PubMed.
- G. N. Rudenskaya, V. A. Isaev, A. M. Shmoylov, M. A. Karabasova, S. V. Shvets, A. I. Miroshnikov and A. B. Brusov, Appl. Biochem. Biotechnol., 2000, 88, 175–183 CrossRef CAS.
- S. K. Agrawal, N. Sanabria-DeLong, G. N. Tew and S. R. Bhatia, Macromolecules, 2008, 41, 1774–1784 CrossRef CAS.
- Y. Li, R. Tong, H. Xia, H. Zhang and J. Xuan, Chem. Commun., 2010, 46, 7739–7741 RSC.
- T. Yamaguchi, M. Nomura, T. Matsuoka and S. Koda, Chem. Phys. Lipids, 2009, 160, 58–62 CrossRef CAS PubMed.
- F. Bordi, C. Cametti, S. Sennato and D. Viscomi, J. Colloid Interface Sci., 2006, 304, 512–517 CrossRef CAS PubMed.
- C. F. Thomas and P. L. Luisi, J. Phys. Chem. B, 2005, 109, 14544–14550 CrossRef CAS PubMed.
- P. Carrara, P. Stano and P. L. Luisi, ChemBioChem, 2012, 13, 1497–1502 CrossRef CAS PubMed.
- A. San Juan, D. Letourneur and V. A. Izumrudov, Bioconjugate Chem., 2007, 18, 922–928 CrossRef CAS PubMed.
- L. Chen, X. Zhigang, J. Hu, X. Chen and X. Jing, J. Nanopart. Res., 2007, 9, 777–785 CrossRef CAS.
- S. K. Agrawal, N. Sanabria-DeLong, S. K. Bhatia, G. N. Tew and S. R. Bhatia, Langmuir, 2010, 26, 17330–17338 CrossRef CAS PubMed.
- T. Fujiwara and Y. Kimura, Macromol. Biosci., 2002, 2, 11–23 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra00835b |
| This journal is © The Royal Society of Chemistry 2015 |