
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile preparation of nanostructured copper-coated carbon microelectrodes for amperometric sensing of carbohydrates†
D.
Riman
a,
Z.
Bartosova
a,
V.
Halouzka
b,
J.
Vacek
c,
D.
Jirovsky
a and
J.
Hrbac
*d
aDepartment of Analytical Chemistry, Palacky University, Faculty of Science, 17. Listopadu 12, CZ-771 46 Olomouc, Czech Republic
bDepartment of Physics and Materials Engineering, Faculty of Technology, Tomas Bata University in Zlin, Nam. T.G. Masaryka 275, 76001 Zlin, Czech Republic
cDepartment of Medical Chemistry and Biochemistry, Faculty of Medicine and Dentistry, Palacky University, Hnevotinska 3, 775 15 Olomouc, Czech Republic
dDepartment of Chemistry, Masaryk University, Kamenice 5, Brno 625 00, Czech Republic. E-mail: jhrbac@atlas.cz
First published on 26th March 2015
Abstract
We report a novel method for fabricating nanostructured copper-coated carbon cylindrical fiber microelectrodes and show the high efficiency of these electrodes in carbohydrate non-enzymatic and label-free amperometric sensing in both batch and flow-detection arrangements.
Carbohydrates are generally regarded as difficult-to-analyze compounds, since they lack suitable chromophores, fluorophores or substituents required for direct detection by spectrophotometric or fluorimetric devices. Under specific conditions, carbohydrates can be measured electrochemically after labelling with electroactive markers.1,2 Of the direct electrochemical methods, pulsed amperometric detection (PAD) at platinum or gold electrodes in alkaline media has attracted a lot of attention as a method that provides a sensitive response in carbohydrate analysis. The method is based on the application of a complex, multistep potential waveform to noble-metal (predominantly gold) working electrodes. This approach effectively solves the problems associated with electrode surface fouling that occurs rapidly when working with simple direct-current methods of carbohydrate oxidation. Although PAD is already a firmly established approach for carbohydrate detection at least for HPLC applications, the constant potential detection mode at metal electrodes offers several advantages over PAD. Apart from its inherently simpler instrumentation, it should, in principle, provide higher sensitivity, because all other parameters being equal, noise should be minimized when the applied potential is constant.3
Therefore, the constant-potential detection of carbohydrates is being intensely studied, with copper as one of the materials of choice due to the specific catalysis of carbohydrate oxidation based on CuII/CuIII redox cycling in surface copper oxides. In the last decade, electrodes modified with copper-based nanomaterials were introduced as efficient devices in carbohydrate analysis, particularly in glucose sensing. Non-enzymatic glucose sensors, based on the combination of copper/copper oxide nanoparticles and carbon (carbon nanotubes and graphene) nanomaterials are part of current research trends. These devices achieve micromolar to submicromolar detection limits in 0.1 M NaOH (1.0 μmol L−1 for Cu nanocubes/MWCNT4 and 0.2 μmol L−1 for irregular Cu nanoparticles/graphene5) with response times in seconds. In general, the nanostructuring of catalytic electrode surfaces offers a combination of increased electroactive surface area with a decrease in the potentials of surface redox transitions.6 When applied as sensors, electrodes modified with nanomaterials frequently allow for higher sensitivity and a broader concentration range of analytes in comparison to their bulk counterparts.7 Nevertheless, when used it should be kept in mind that some gradual loss of outer material via dissolution at the electrode/solution interface may occur at such electrodes depending on the nanostructured film thickness, possibly leading to a lower response stability.
Despite the known benefits of employing metal electrodes in the electroanalysis of carbohydrates, a limited number of papers cover the detection of carbohydrates by constant potential amperometry in flow systems and only a few papers have been devoted to the high pressure liquid chromatography with electrochemical detection (HPLC-ED) of carbohydrates based on copper electrodes.8–12
In our previous works, we have reported that carbon fiber microelectrodes (CFMEs) can be utilized as highly sensitive electrochemical detectors, especially in HPLC.13–16 Thereby, we developed a facile technique enabling CFMEs to be modified with nanostructured copper layers. The dimensions of a carbon fiber (3–4 mm in length, 7 μm in diameter) enable the direct insertion of the active electrode tip into a narrow-bore capillary even after the modification with copper. The advanced mechanical properties of carbon fibers grant the resulting sensing element excellent mechanical stability, superior to other electrode materials of similar size and shape, e.g. extremely fragile copper microwires. The copper-modified CFME exhibits high performance in carbohydrate electrooxidation-based sensing, as tested in the amperometric, flow injection analysis (FIA) and HPLC detection of selected mono-, di- and oligosaccharides.
Preparation of copper-coated carbon fiber microelectrode
The procedure for CFME modification by copper is based on a methodology15 previously described by ourselves for the preparation of nanostructured silver layers, i.e. electrodeposition of the material formed by the dissolution of the anode. The formed anode-derived material is transferred towards the cathode by movement through the interelectrode space originating from the combined action of migration, diffusion and convection induced by thermal and density gradients. Reductive deposition occurs at the cathode. Specifically, copper wire was anodized in a two-electrode cell containing ultrapure water as the medium while the CFME was connected as the cathode, the interelectrode distance being 1 cm. Due to the low conductivity of ultrapure water, relatively high potentials (10–30 V) had to be employed to induce copper anodic dissolution. Following optimization experiments, a potential of 15 V was applied to the electrodes for 20 min. These conditions represent a right compromise between the analytical performance of the modified CFME and stability of the modifying layer in FIA and HPLC arrangements (see below). We preclude that copper electrodeposition proceeds by a similar mechanism to that suggested for silver. It is to be expected that the potential is sufficient to induce a two-electron oxidation of copper (eqn (1)).17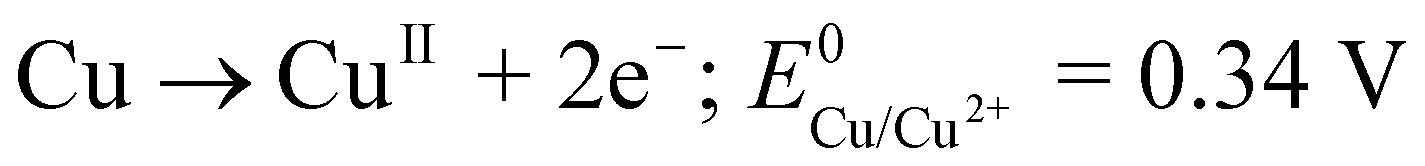 | (1) |
Cu(II) readily reacts with OH− available from the autoprotolysis of water to give Cu(OH)2 (eqn (2))
| CuII + 2H2O → Cu(OH)2 + 2H+. | (2) |
The solubility product of Cu(OH)2,
| Ks = 2.2 × 1020, | (3) |
| H+ + e− → 1/2H2 | (4) |
The hydrogen gas formed may contribute towards keeping the copper layer in the reduced state. The copper-coated CFMEs were examined by scanning electron microscopy (SEM), revealing that the deposit is a randomly oriented Cu nanostructured network (Fig. 1A and B). The elemental composition of the deposit was verified by energy dispersive X-ray (EDX) spectroscopy (Fig. 1C). In addition to the carbon peak originating from the carbon substrate, only copper features and a small peak of oxygen, presumably formed as an artefact due to manipulating with the copper-coated CFME in an air atmosphere, were found.
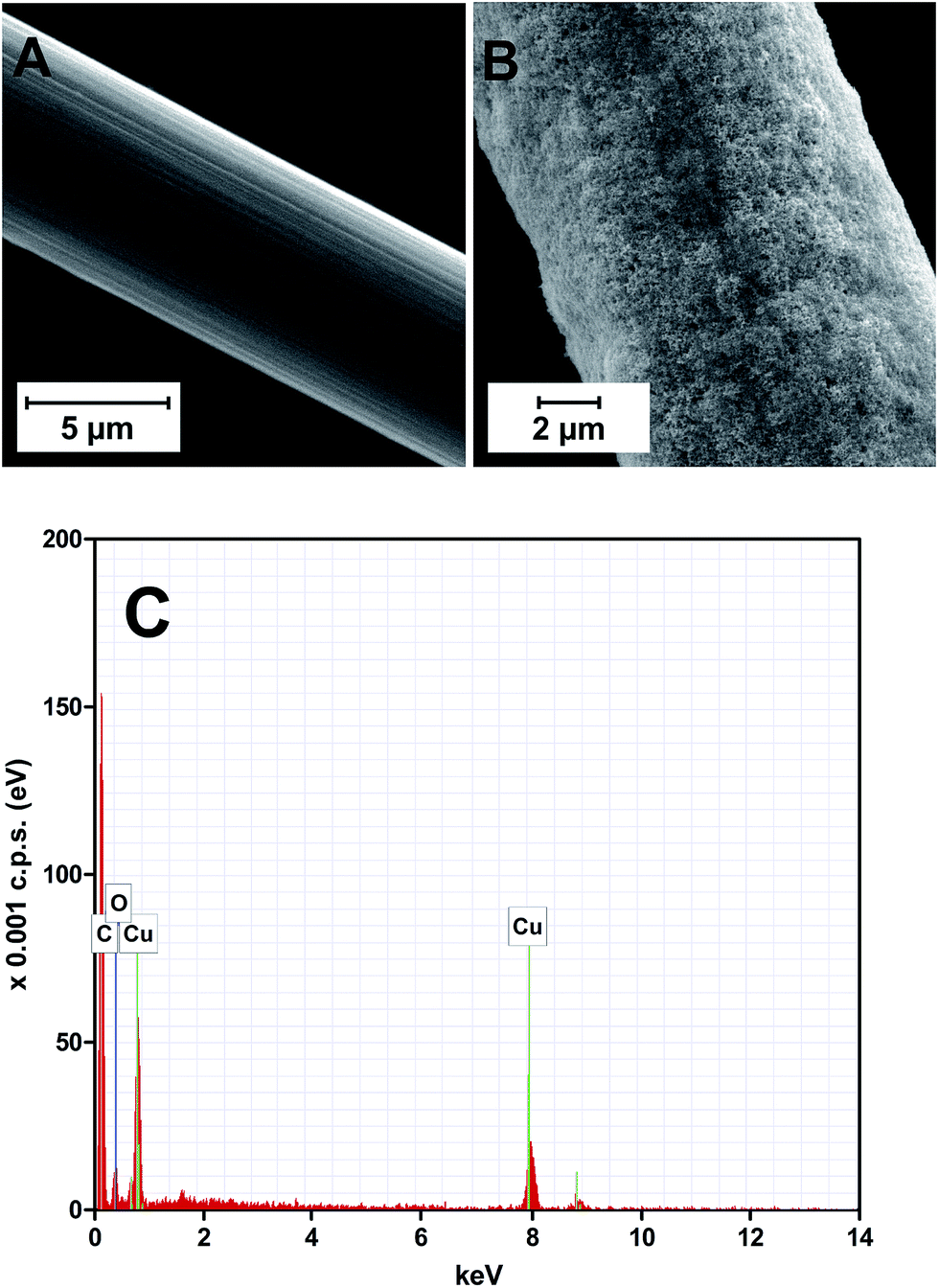 | ||
| Fig. 1 SEM images of (A) bare carbon fiber, (B) copper-coated carbon fiber; (C) EDX spectrum of (B). | ||
Cyclic voltammetric (CV) record of the copper-coated CFME, for comparison shown along with the CV of a copper wire electrode recorded under identical conditions, contains all copper redox transitions (peaks I and II in Fig. 2) and is identical to the CV records reported in the literature.21,22
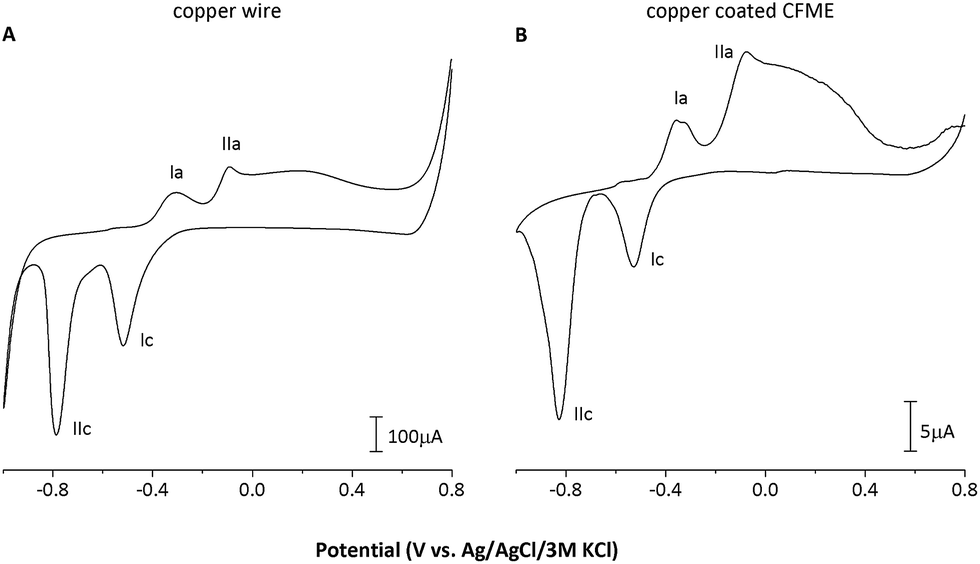 | ||
| Fig. 2 Cyclic voltammograms of (A) copper wire electrode and (B) copper-coated CFME recorded in 0.01 mol L−1 NaOH, scan rate = 100 mV s−1, Einit +0.8 V. Ia: Cu0 → CuI; IIa: CuI → CuII; Ic: CuII → CuI; IIc: CuI → Cu0. | ||
The copper wire electrode used, having an almost 200-fold higher geometric area than the copper-coated CFME (2.4 × 10−5vs. 1.4 × 10−7 m2), provided only ca. 20-fold higher currents for the observed surfacial redox transitions, i.e. approx. 10-fold higher current densities were achieved on the CFME due to electrodeposition (Fig. 2).
The performance of copper coated CFMEs in carbohydrate sensing
The performance of copper electrodes in carbohydrate sensing depends heavily on the pre-oxidation of the copper electrode to form catalytically active surface copper oxides. Various anodisation procedures were suggested in the literature, leading to differently sensitive surfaces; probably the most sensitive electrodes were reported by Kano et al.,8 who oxidised a polished copper electrode by the action of hot air (100 °C, 3 h) and employed these electrodes in the detection of a mixture of sugars in 0.15 mol L−1 NaOH at 0.45 V vs. Ag/AgCl after HPLC separation. The drawback of these electrodes was the requirement of 2 hours of stabilization of the background current. For copper-coated CFMEs, satisfactory results were achieved using a simpler and less time-consuming procedure, i.e. anodisation at +700 mV vs. Ag/AgCl for five minutes. In this procedure, a complete transformation accompanied by a morphology change to cube-shaped, 50–400 nm sized copper oxide particles occurs (Fig. 3). | ||
| Fig. 3 SEM of copper-coated CFME after anodisation in 0.01 mol L−1 NaOH at +0.7 V vs. Ag/AgCl for 5 min. | ||
Experiments demonstrating the performance of the copper nanostructured CFMEs in the amperometric sensing of glucose were then performed, employing working potentials between 500–800 mV vs. Ag/AgCl. Optimum performance was observed between 600–700 mV, though for the applied potential of 500 mV, both the electrode response and background were lowered proportionally. On the other hand, for the applied potential of 800 mV (trace not shown) the sensitivity of the nanostructured CFMEs towards glucose remained similar to that observed at 700 mV, but the baseline current was markedly elevated. The nanostructured CFMEs therefore provide usable analytical signals when operated between 500–700 mV vs. Ag/AgCl, which is clearly shown in Fig. 4.
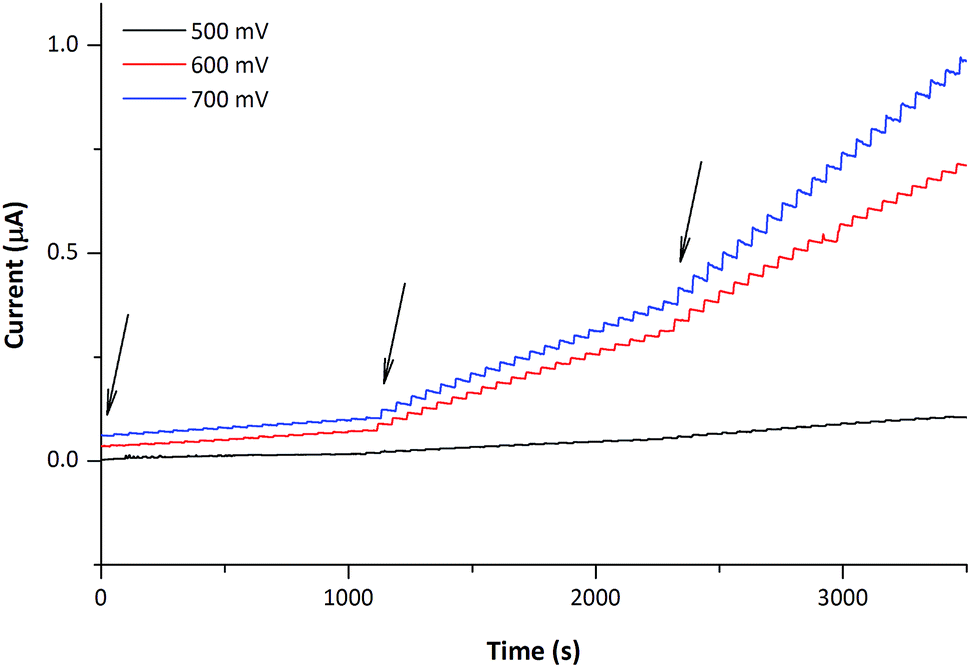 | ||
| Fig. 4 Amperometric traces of glucose at different applied working potentials. 0.01 mol L−1 NaOH was used as supporting electrolyte, additions (series indicated by arrows) led to the following increases in glucose concentration: 20-times 5 × 10−6, 20-times 1 × 10−5 and 20-times 2 × 10−5 mol L−1, stirred solution (300 r.p.m.). | ||
Rapid response times, high surface area, radial diffusion and the guaranteed electrical contact with the carbon fiber substrate ensured by the electrochemical nature of the nanostructures' formation lead to increased analytical usability of fabricated nanostructured copper-coated CFMEs. Therefore, we incorporated the sensor, treated according to the procedure described above, into a FIA/HPLC detection flow-cell, described in our previous works.13,14 FIA was tested at various flow rates (15–200 μL min−1), nevertheless, using our instrumentation, similar values of LODs were found (Table 1). For illustration, the FIA record of lactose over a broad concentration range is shown in Fig. 5A.
| Saccharide | LOD (μM L−1) | LOQ (μM L−1) | R 2 (peak area) | R 2 (peak height) | Sensitivity (A M−1 m−2) |
|---|---|---|---|---|---|
| a Data for dextran in μg mL−1. b Sensitivity in A g−1 m−2; 0.025 mol L−1 NaOH as mobile phase; flow rate = 200 μL min−1; working potential 700 mV. | |||||
| Xylose | 2.85 | 9.51 | 0.9985 | 0.9947 | 238 |
| Fructose | 1.01 | 3.36 | 0.9932 | 0.9921 | 514 |
| Galactose | 1.27 | 4.23 | 0.9989 | 0.9998 | 548 |
| Glucose | 2.63 | 8.77 | 0.9997 | 0.9982 | 267 |
| Sucrose | 1.19 | 6.40 | 0.996 | 0.9956 | 51 |
| Maltose | 0.88 | 2.90 | 0.986 | 0.9812 | 110 |
| Lactose | 0.35 | 1.16 | 0.9919 | 0.9822 | 368 |
| Raffinose | 0.84 | 2.81 | 0.9934 | 0.9947 | 54 |
| Dextrana | 0.39a | 1.30a | 0.9995 | 0.9788 | 744b |
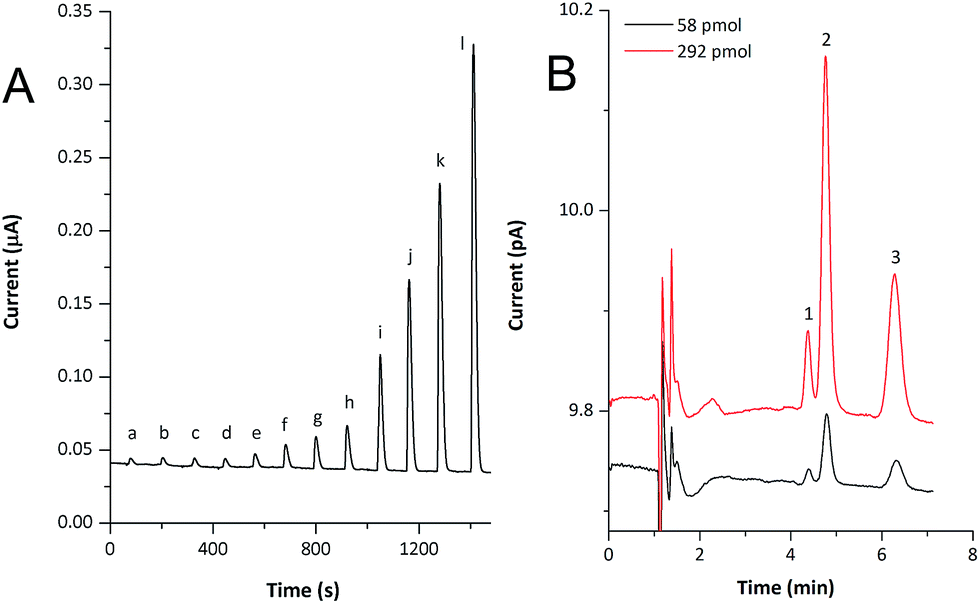 | ||
| Fig. 5 FIA records of lactose (A) and HPLC chromatogram (B) of selected disaccharides obtained using nanostructured copper-coated CFME as amperometric sensor. Flow rate: 15 μL min−1 (for FIA) and 200 μL min−1 (for HPLC), working potentials 700 mV (for FIA) and 500 mV (for HPLC) vs. Ag/AgCl. Legend: a – 2.5 × 10−5; b – 5 × 10−5; c – 7.5 × 10−5; d – 1 × 10−4; e – 2.5 × 10−4; f – 5 × 10−4; g – 7.5 × 10−4; h – 1 × 10−3; i – 2.5 × 10−3; j – 5 × 10−3; k – 7.5 × 10−3; l – 1 × 10−2 mol L−1 of lactose; 1 – sucrose, 2 – lactose, 3 – maltose. | ||
The response stability of the nanostructured electrode in FIA mode was tested for lactose. For this purpose, five consecutive injections of lactose solution were carried out at 0, 0.5, 1, 2 and 3 h from the beginning of the test. After overnight break, the test was continued at 12, 13 and 14th hour. For details, see Fig. S3 and corresponding part of the ESI.† The R.S.D. within individual test runs ranged from 1.6–7.2%. The interelectrode reproducibility was 20%, which was deemed satisfactory considering differences in surface areas and state of the individual carbon fibers and a difficulty to control their exact lengths in the assembled CFMEs.
Finally, the performance of the detector in an HPLC setup was investigated using a model mixture of three disaccharides (sucrose, lactose and maltose). For the separation we used an HPLC column, which is, unlike the majority of silica-based sorbents, alkali-resistant, and thus no extra post-column addition of concentrated hydroxide solution is needed. This way, the detection of carbohydrates can be accomplished directly in the mobile phase and the necessity of other additional instrumentation (such as a pulseless pump for post-column alkalinisation of the mobile phase) is eliminated. Similarly to the FIA experiment, the sensor exhibited a rapid response, very short reacquisition times and good baseline stability within runs. Highly stable, low-noise backgrounds were achieved, especially when the CFME detector was operated at a low applied potential. The HPLC analysis of disaccharides (292 or 58 pmol per column) is shown in Fig. 5B.
Conclusions
Carbon fiber microelectrodes coated with a copper layer were prepared using the above novel electrodeposition method. Following electrochemical transformation of the copper layer to copper oxide, the resulting device was shown to be an efficient non-enzymatic carbohydrate sensor, exhibiting low detection limits and a wide linear concentration range, comparable to the best copper-based sensors, e.g. a sensor utilizing a graphene–copper composite.5 Short response time, small dimensions and high stability favour the use of the sensor in FIA and HPLC systems. The results presented here could be the starting point for the further development of novel nanostructured microelectrodes for not only the analysis of carbohydrates in solution, but also sensing their binding to proteins, i.e. the trace detection of glycoproteins or monitoring of non-enzymatic glycation processes.Acknowledgements
The research was supported by the Grant Agency of the Czech Republic, projects P206/12/0796 (J.H.) and 14-08032S (J.V.). The authors wish to thank Mr Jan Kostejn, BSc. for his assistance with amperometric measurements, Dr Milan Vujtek for SEM imaging, and Mr Ben Watson-Jones, MEng for language correction.Notes and references
- E. Palecek, M. Bartosik, V. Ostatna and M. Trefulka, Chem. Rec., 2012, 12, 27–45 CrossRef CAS PubMed.
- E. Palecek, V. Ostatna, M. Trefulka, M. Bartosik, H. Cernocka, K. Kurzatkowska and M. Zivanovic, Proc. IEEE Sens., 2010, 833–836 Search PubMed.
- R. P. Baldwin, in Carbohydrate Analysis by Modern Chromatography and Electrophoresis, ed. Z. El Rassi, Elsevier, Amsterdam, 2002, pp. 947–959 Search PubMed.
- J. A. Yang, W. D. Zhang and S. Gunasekaran, Biosens. Bioelectron., 2010, 26, 279–284 CrossRef CAS PubMed.
- J. Luo, S. S. Jiang, H. Y. Zhang, J. Q. Jiang and X. Y. Liu, Anal. Chim. Acta, 2012, 709, 47–53 CrossRef CAS PubMed.
- W. J. Plieth, J. Phys. Chem., 1982, 86, 3166–3170 CrossRef CAS.
- C. Zhu, G. Yang, H. Li, D. Du and Y. Lin, Anal. Chem., 2015, 87, 230–249 CrossRef CAS PubMed.
- K. Kano, K. Takagi, K. Inoue, T. Ikeda and T. Ueda, J. Chromatogr. A, 1996, 721, 53–57 CrossRef CAS.
- P. F. Luo, S. V. Prabhu and R. P. Baldwin, Anal. Chem., 1990, 62, 752–755 CrossRef CAS.
- L. Nagy, G. Nagy and P. Hajos, Sens. Actuators, B, 2001, 76, 494–499 CrossRef CAS.
- S. V. Prabhu and R. P. Baldwin, Anal. Chem., 1989, 61, 852–856 CrossRef CAS.
- S. A. Wring, A. Terry, R. Causon and W. N. Jenner, J. Pharm. Biomed. Anal., 1998, 16, 1213–1224 CrossRef CAS.
- Z. Bartosova, D. Jirovsky, D. Riman, V. Halouzka, M. Svidrnoch and J. Hrbac, Talanta, 2014, 122, 115–121 CrossRef CAS PubMed.
- Z. Bartosova, D. Riman, P. Jakubec, V. Halouzka, J. Hrbac and D. Jirovsky, Sci. World J., 2012, 295802 Search PubMed.
- V. Halouzka, P. Jakubec, L. Kvitek, V. Likodimos, A. G. Kontos, K. Papadopoulos, P. Falaras and J. Hrbac, J. Electrochem. Soc., 2013, 160, B54–B59 CrossRef CAS PubMed.
- P. Jakubec, M. Bancirova, V. Halouzka, A. Lojek, M. Ciz, P. Denev, N. Cibicek, J. Vacek, J. Vostalova, J. Ulrichova and J. Hrbac, J. Agric. Food Chem., 2012, 60, 7836–7843 CrossRef CAS PubMed.
- P. Patnaik, Handbook of inorganic chemicals, McGraw-Hill, New York, 2003 Search PubMed.
- V. Fleury, J. N. Chazalviel, M. Rosso and B. Sapoval, NATO Adv. Sci. Inst. Ser., Ser. E, 1990, 188, 304–305 Search PubMed.
- K. Kondo, Morphological evolution of electrodeposits and electrochemical processing in ULSI fabrication and electrodeposition of and on semiconductors IV: Proceedings of the international symposia, Electrochemical Society, Pennington, NJ, 2005 Search PubMed.
- S. Yagi, H. Nakanishi, T. Ichitsubo and E. Matsubara, J. Electrochem. Soc., 2009, 156, D321–D325 CrossRef CAS PubMed.
- L. D. Burke, M. J. G. Ahern and T. G. Ryan, J. Electrochem. Soc., 1990, 137, 553–561 CrossRef CAS PubMed.
- C. H. Pyun and S. M. Park, J. Electrochem. Soc., 1986, 133, 2024–2030 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical and reagents; carbon fiber microelectrode fabrication; modification of carbon fiber microelectrode with copper; amperometric detector layout; scanning electron microscopy of CFMEs and EDX spectroscopy; amperometry/FIA/HPLC instrumentation and analytical conditions, response stability testing. See DOI: 10.1039/c5ra00831j |
| This journal is © The Royal Society of Chemistry 2015 |