
Synthesis and characterization of new hydrodesulphurization Co–Mo catalysts supported on calcined and pyrolyzed bone
Abstract
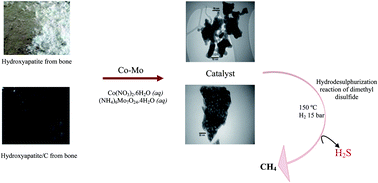
A series of cobalt–molybdenum catalysts supported on calcined bone (hydroxyapatite, HAP) and pyrolyzed bone (hydroxyapatite–carbon, HAP–C) have been prepared by an impregnation method. The catalysts were characterized by XRD, FT-IR spectroscopy, UV-Vis spectroscopy, TPR, TPD of CO2, BET, FE-SEM, EDX and TEM techniques. Hydrodesulphurization (HDS) experiments were carried out in a fixed-bed reactor at 15 bar hydrogen pressure, liquid hourly space velocity (LHSV) of 4.5 h−1, a volumetric hydrogen/feed ratio of 175 NL L−1 and 310 °C. HDS reactions of dibenzothiophene (DBT), dimethyl disulfide (DMDS) and industrial naphtha over the synthesized catalysts were investigated. The Co–Mo/HAP–C and Co–Mo/HAP catalysts displayed a satisfactory performance comparable with the commercial catalyst. Co–Mo/Al2O3 and Co–Mo/HAP catalysts were tested for hydrodesulphurization of DMDS, and Co–Mo/HAP showed a higher activity than the commercial catalyst (Co–Mo/Al2O3) and lowered the sulfur content of the feedstock from 2500 ppm to 9 ppm.
 Please wait while we load your content...
Please wait while we load your content...

