
Microwave induced plasma desorption ionization (MIPDI) mass spectrometry for qualitative and quantitative analysis of preservatives in cosmetics
Zhongjun Zhaoa and
Yixiang Duan*b
aCollege of Chemistry, Sichuan University, Chengdu, PR China
bResearch Center of Analytical Instrumentation, Key Laboratory of Bio-resource and Eco-environment, Ministry of Education, College of Life Sciences, Sichuan University, 29 Wangjiang Road, Chengdu, 610064, PR China. E-mail: yduan@scu.edu.cn; Fax: +86-028-85418180; Tel: +86-028-85418180
First published on 14th April 2015
Abstract
Ambient ion sources for mass spectrometry have been frequently reported in the past 10 years. The most attractive features of these ion sources are that they are fast and an easy way to analyze various samples. The microwave induced plasma desorption ionization source, the MIPDI source, is one among them. In this study, the qualitative and quantitative behavior of the MIPDI source has been demonstrated by analyzing the preservatives in cosmetics for the first time. The detection limit for the preservatives is as low as pg mm−2. The relative standard deviation of continuous analysis is 5.25%. Preservatives in commercial cosmetics samples were successfully detected including those in facial cream, sunscreen and moisturizer. The fast screening capability of the MIPDI source is proved. Five commercial samples were successfully classified into two groups within 5 minutes according to the added preservatives. The ability to quantitatively analyze the preservatives in commercial cosmetics was also investigated. The standard adding and calibration curve methods were used in the quantitation process. The results showed that the quantitative analysis accuracy of MIPDI-MS is −49% and −66% for a liquid state sample and solid state sample respectively, i.e. semi-quantitation is possible. A conceptual experiment was also conducted to validate the accurate quantitative analysis capability of MIPDI-MS. The concentration of caffeine in cosmetic matrices was quantified satisfactorily with no sample pretreatment, with an RSD value of 5.5% and accuracy of 3.6%. The approaches established in this work indicate that the MIPDI source is a promising tool in future applications where rapid qualitative and quantitative analysis is needed.
Introduction
Preservatives, which are typically antibacterial agents, are added into cosmetics because the contents of cosmetics are usually organic substances, which are suitable for microbial growth. Also, antioxidants are used in order to retain the effectiveness of the functional substance. Many antibacterial agents contain chlorine or bromine to suppress the growth of microorganisms.1 The halogen is usually allergenic and/or poisonous to the human body. To control the use of these preservatives, the additives to cosmetics are strictly limited by the Ministry of Health of the P.R.C. National Hygienic Standard for Cosmetics (NHSC). Over 1200 forbidden additives and 56 permitted preservatives with their maximum dosages are listed by the NHSC to ensure the contact safety of products. Excessive adding of cosmetic preservatives will increase the potential hazard to health. These substances may lead to acute oral toxicity, acute dermal toxicity, dermal irritation/corrosion, skin sensitization, skin phototoxicity, cellular chromosome aberration, cell gene mutation, teratogenicity and even carcinogenicity. Thus, the analysis of preservatives in cosmetics is a socially relevant and challenging activity. The NHSC has drafted a series of regulations and testing protocols. The recommended analysis method from the NHSC is either high performance liquid chromatography (HPLC) or gas chromatography (GC), the most widely used techniques all over the world. These methods provide accurate and plentiful information on the substance of interest. However, reliable data requires appropriate chromatography conditions and long separation times. Furthermore, sample manipulation takes much time and effort. Due to the complex content of cosmetics, an extraction process must be done before chromatography separation is feasible. It has so far been impossible to establish a fast screening and classifying method based on the above techniques. It is therefore worthwhile to search for an alternative method that would avoid the shortcomings of the traditional analysis technologies.The newly emerging technique of ambient desorption mass spectrometry has captured the attention of analysts. The analysis process can be much shortened with the use of a mass spectrometer equipped with an ambient desorption ionization source. Ambient desorption ionization sources for mass spectrometry have been studied for years, extending the use of the power of the mass spectrometer. Sample pre-treatments before testing are minimal to none when using ambient desorption ionization sources. Molecules in relatively raw matrices (solid state, liquid state or gaseous state) can be directly ionized, i.e. direct analysis of samples is feasible. This means that in situ analyzing can be realized for workers who want analytes to be kept in their original chemical2–4 and biological5–9 environments. For these reasons, ambient desorption ionization sources have been applied to the problems of directly analyzing food additives,10,11 for carcinogenic aromatic amines in textiles,2,3 for pharmaceuticals12–14 and even for metabolites in live cells or tissues.15–17 The ambient desorption ionization sources demonstrated a great reliability for qualitative analysis in these applications. At the same time, the ambient desorption ionization sources provided as low as pg mm−2 detection limit.18,19 The low detection limit makes the ambient desorption ionization sources ideal tools to detect trace substances in complex matrices. Benefiting from the direct ionization of molecules from their native environments, the whole analysis time is short enough (more than 1 sample per second) to be applied in situations where a high through-put test is demanded. Besides, the mass spectra resulting from the use of ambient ionization sources are usually clear without fragment ions. Molecular ions can be easily identified from the spectra. The ability of qualitatively saying yes or no to the presence of a certain substance is quite effective, which means fast screening and classification can be achieved. Owing to these very desirable features, many kinds of ambient ionization sources have been developed. These ion sources are mainly divided into two species, ESI based and plasma based. The recently introduced microwave induced plasma desorption ionization (MIPDI) source19–21 is one among many plasma based ambient desorption ionization sources18,22–24 for mass spectrometry. In a MIPDI source, the discharge gas (argon or helium) is ionized or excited by resonating with microwave power. The produced argon/helium ions and high energy neutral species quickly react with the neutral species in air, forming secondary ions and metastable state neutrals. These relatively stable ions and metastable state neutrals (reactants) are ejected from the MIPDI source to the surface of the sample with the flow of the plasma jet. Once these reactants contact the target molecules on the surface of the sample, proton transfer reactions or Penning ionization happens. The ionized target molecules subsequently enter the ambient pressure interface and are detected by the mass spectrometer. In our previous work, the MIPDI source has been successfully applied for the qualitative detection of the active ingredients of pharmaceuticals. The MIPDI source showed good tolerance to a complex matrix of tablets and ointments and great reliability in qualitatively showing the existence of target molecules. However, the existing research of the MIPDI source has simply examined its qualitative ability; no report has systematically emphasized the qualitative and quantitative direct analysis of cosmetic preservatives or any similar substances without any kind of sample treatment. It is of potentially great value if accurate quantitative analysis and potential fast screening capability of the MIPDI source can be developed to meet the demands for commercial cosmetic preservatives detection.
To evaluate the application of the MIPDI source in the fast detection and classification of preservatives in cosmetics, 6 of the most commonly used preservative compounds (Table 1) were selected as representatives of the class as a whole. The detection limits of the MIPDI source to those preservatives were examined. Both liquid and solid state (ointment) cosmetic samples were included in performing the fast screening experiments by the MIPDI source, covering the categories of sunscreen, facial cream and moisturizer. The fast classification capability of the MIPDI source was examined by a blind classification of 5 commercially available cosmetics according to the added preservatives. The quantitative analysis capability of the MIPDI source had not been studied. The ability has been commonly investigated by workers using ambient ionization sources.25–29 To remedy this oversight, the quantitative analysis capability of the MIPDI source was systematically examined using two methods, the standard adding method and the calibration curve method. The isotopically labeled standard method was also verified using a proof-of-concept experiment to study the accurate quantitative analysis ability of the MIPDI source. Finally, the result was referenced by the NHSC accepted HPLC protocols.
| Compound | M.W. | Selected ion (m/z) | D.L. | Solution (w/w) | RSD |
|---|---|---|---|---|---|
| Hexamethylenetetramine | 140.2 | 141.0 | 5.2 pg mm−2 | 1.04 ppb | 5.2% |
| 4-Phenylphenol | 170.2 | 170.1 | 5.7 pg mm−2 | 1.14 ppb | 12.4% |
| 2-Methyl-4-isothiazolin-3-one | 115.2 | 116.0 | 3.0 pg mm−2 | 0.60 ppb | 6.7% |
| Climbazole | 292.8 | 293.1 | 5.7 pg mm−2 | 1.14 ppb | 8.9% |
| Hexetidine | 339.6 | 354.6 | 4.3 pg mm−2 | 0.86 ppb | 5.5% |
| 2-Benzyl-4-chlorophenol | 218.7 | 217.2 | 400 pg mm−2 | 80.0 ppb | 8.4% |
Experimental section
Chemicals and reagents
Preservative samples (analytically pure) were purchased from Sigma-Aldrich (Germany). Methanol (HPLC grade) was obtained from Honeywell. D3-caffeine (3H substituted by 3D, M.W. 197) was purchased from Quandao Company (China). Other chemicals were products of Sinopharm Chemical Reagent Company (China) and were used without further purification throughout the experiment. Ultra purified water (18 MΩ cm−3) was produced using a UP water purification system (Youpu Company, China). The discharge gas, argon (99.999%), was purchased from ShenQi Gas Company (Chengdu, China). Cosmetic samples, including sunscreen, facial cream and moisturizer, were purchased locally. Glass slides (China, 25.4 mm × 76.2 mm) were the product of the Sail Brand Company.MIPDI source configuration
The MIPDI source is a type of surface wave generator called a surfatron. The source has been described by our group20,21 previously and detailed information about the experimental set-up can be found there. The resonance cavity is in a brass cylinder. In the center of the cavity, a quartz tube is mounted axially. The discharge gas flow passes through the quartz tube. In this device, microwaves travel along the surface of the quartz tube in the cavity, forming an argon plasma inside the quartz tube. The quartz tube is of 0.8 mm i.d., 6 mm o.d., and is 200 mm long. The plasma is formed along the surface of the inside of the quartz tube and is a quasi-conical jet. The microwave induced plasma jet outside of the quartz tube is needle-like. The color of the plasma is bright purple. The resonation cavity was tuned to ensure it worked in its best configuration i.e. at the lowest reverse power. The MIPDI source was installed on a rotating stage. The rotating stage was mounted on a 3D moving stage (X–Y–Z) in front of the mass spectrometer ion transfer capillary (Scheme 1). Thus, the whole MIPDI source can be moved in 3 dimensions and 1 angle. The sample stage was fixed at the same level as the ion transfer capillary through which the ions were transported to the mass analyzer. Overall, the plasma jet is at a 45° angle to the sample stage and at a 135° angle to the ion transfer capillary. The microwave power generator (2.45 GHz, 150 W max, Nanjing Yanyou Electronic Science and Technology Co. Ltd.) was set to 6 V, equal to 70 watts. Argon was used as the discharge gas with a flow rate of 850 mL min−1. The flow was controlled by a mass flow controller (D07-19B, Beijing Sevenstar Electronics Co. Ltd.). Under the above conditions, the plasma jet was about 5 mm out of the MIPDI source. | ||
| Scheme 1 Overview of the MIPDI source in its experimental configuration. | ||
Mass spectrometer conditions
A 3D ion trap mass spectrometer (LCQ Fleet, Thermo Fisher Scientific; San Jose, CA) was used throughout this work. All of the voltage settings of the mass spectrometer were tuned and calibrated with the tuning method using ESI as the ion source. The tube lens was manually set to 80 volts and the ion transfer capillary was set to 30 volts to satisfy the mass to charge ratio range of interest, which ranged from m/z 100 to m/z 500. Target molecules were ionized while desorbing, so the MIPDI process did not need any method for removing the solvent. As a consequence, the ion capillary temperature was decreased to 175 °C. The mass spectrometer was working in full scan and in positive ion mode (normal scan, above m/z 50) in this study. The spray voltage, auxiliary gas, sweep gas and sheath gas were turned off. The maximum ion injection time was set to 500 ms while the micro scan was twice each full scan. The instrument software “Xcalibur” was used to process the data from the mass spectrometer.Sample treatments and preparations
Both filter paper and glass slides were examined as potential sample holders. Results showed that filter paper gave more background ions. Also, the sample area was hard to confine due to the chromatography phenomenon. That is, the actual sample distribution area is not as large as the solvent ring. Glass slides were therefore used as the sample support system to avoid the shortcomings of filter paper. A 5 μL analyte sample was pipetted onto the glass slide. The pipetted sample quickly spread out, forming a sample ring. The sample solution dispersed evenly on the glass slide surface. The area of the sample ring was about 10 mm2. Experiments were carried out as the methanol vaporized. Two main experiments were performed using the MIPDI source in this study: (1) qualitative analysis of preservatives in commercial cosmetics and (2) quantitative analysis of preservatives in commercial cosmetics. The samples were divided into two types, the solid state and the liquid state. The sample preparation was quite different in those two experiments.For the qualitative analysis of preservatives in commercial cosmetics, the sample preparation was simple. The liquid cosmetic sample was diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) using methanol as the solvent. The diluted solution was pipetted onto the glass slides and analyzed by the MIPDI source. The solid state cosmetic sample, i.e. sun screen (5 mg), was evenly smeared directly onto the glass slide. The doped glass slides were then ready to be analyzed using the MIPDI source.
:1 (v/v) using methanol as the solvent. The diluted solution was pipetted onto the glass slides and analyzed by the MIPDI source. The solid state cosmetic sample, i.e. sun screen (5 mg), was evenly smeared directly onto the glass slide. The doped glass slides were then ready to be analyzed using the MIPDI source.
For the quantitative analysis of preservatives in commercial cosmetics, the sample treatment process depended on the state of the sample and the quantitative analysis method. The standard adding method was used to quantitatively analyze liquid cosmetic samples. The liquid cosmetic was 1:1 (v/v) diluted and the solution was used without any further treatment. The calibration curve method was used to quantitatively analyze solid state cosmetic samples, so the sample was extracted with methanol to prepare a sample stock solution. The extraction routine was the same as recommended by the NHSC. A 1.00 g sample of the cosmetic was dissolved in 10 mL of methanol. The solution was energetically shaken for 15 min. After centrifugation, the mixture was filtered through a 0.45 μm filter membrane (organic phase, Hengxin Company). The filtered solution was used as the sample stock solution, which was ideal for going through the quantitative analysis using the calibration curve method.
Isotopically labeled standards method
The use of an isotopic labeled standards method for the MIPDI quantitative analysis was validated in this work. Due to the lack of an isotopically labeled standard preservative, caffeine was selected as a substitute model preservative in order to do a proof-of-concept experiment. Commercial cosmetics were used as a blank matrix. To produce 125 μg mL−1 of “matrix matched cosmetic samples”, a 2 mL aliquot of cosmetic matrix was added to 20 μL of 12.5 mg mL−1 caffeine solution in methanol. These “matrix matched cosmetic samples” were then spiked with known amounts of D3-caffeine (8.6 mg ml−1) 5 μL, 10 μL, 15 μL and 20 μL. The volume difference was adjusted to the standard by adding 15 μL, 10 μL, 5 μL and 0 μL methanol, respectively, as a reference. The D3-caffeine concentrations were 21.5 μg mL−1, 43.0 μg mL−1, 64.5 μg mL−1 and 86.0 μg mL−1, respectively.Liquid chromatography reference method
To ascertain the quantitative analysis accuracy of the MIPDI source, high performance liquid chromatography (HPLC) was performed as a reference according to the NHSC standards. An HPLC machine (Agilent 1220 Infinity) was equipped with an Agilent zorbax eclipse XDB-C18 4.6*250 mm 5-micron C18 reversed-phase analytical column and a UV diode array detector. The wavelength 280 nm was chosen for the analysis, as recommended by the NHSC. Sample treatment before HPLC was as follows: a 1.00 g sample was placed into a cuvette. A water bath was used to remove the volatile solvent. Samples were diluted to 10 mL with methanol. The solution was shaken for 15 min with ultrasonic extraction. After centrifugation, the solutions were filtered through a 0.45 μm membrane to produce the sample stock solution. The calibration solution was prepared by directly dissolving the target preservatives in methanol. The mobile phase, a solution of 50% 0.05 M sodium dihydrogen phosphate, 35% methanol and 15% acetonitrile was prepared, ignoring the slight volume changes on mixing. Hexadecane trimethylamine chloride was dissolved in this solvent to a concentration of 0.002 M. The pH of the final solution was adjusted to 3.5 with phosphate buffer. During the HPLC analysis, the mobile phase flow rate was set to 1.5 mL min−1 and the column temperature was ambient. A calibration curve was constructed with 3.5 μL of 2-methyl-4-isothiazolin-3-one (MIT, standard preservative, 100 mg L−1, 500 mg L−1 and 1000 mg L−1).Safety considerations
Microwave radiation can be hazardous to people and may lead to pathological changes. Furthermore, electrical shock may happen when igniting the plasma with a slender metal wire. Precautions such as aluminum foil clothing, safety glasses, and electrically insulating gloves should be worn.Results and discussion
Analytical performance of MIPDI-MS
The detection limits of 6 preservatives were investigated with optimized source parameters (discharge gas flow rate 850 sccm, microwave input power 6 V) by MIPDI-MS. These samples were pure preservatives dissolved in methanol. The concentration of these samples ranged from 30 to 70 mg mL−1. To discover the detection limit, each preservative, contained in 6 identical samples, was tested six times repeatedly. Abnormal values were evaluated with the 4d inspection method.30 The detailed results of the detection limits and basic information about the preservatives are summarized in Table 1. The ions selected for calculating the detection limits were the protonated molecular ion or the molecular ion. The detection limits of the MIPDI-MS of the selected preservatives were as low as 3.0 pg mm−2 and in the range of 3.0–5.7 pg mm−2 except in the case of climbazole. The relatively poor detection limit of climbazole (400 pg mm−2) was due to its high vaporization point. This high vaporization point makes climbazole harder to be desorbed/ionized. Overall, the performance of the MIPDI source is remarkable and can easily fulfill the analysis requirements of preservatives for cosmetics under the requirements of the NHSC.To evaluate the accuracy of the MIPDI source when used in the detection of preservatives, it is necessary to check the relative standard deviation of the individual test. An acceptable RSD is a basic requirement for the possibility of further quantitative analysis. In an RSD test experiment, MIT was tested repeatedly with a set of six identical samples, as described previously. Integrated peak areas (ion current) of the set of samples were used to evaluate the deviation among the tests. Fig. 1 is the selected ion current of MIT (m/z 116 ± 0.5) of the full scan total ion current of the 6 individual tests. The RSD of the 6 individual MIT tests was 6.7%. The RSD of the other preservatives are listed in Table 1. The relatively less satisfactory RSD of 4-phenylphenol (m/z 170) is the result of the unstable formation process of dimers (m/z 338, 339). The formation of dimers depends heavily on the ionization temperature. However, due to the open ion source, the air flow in the laboratory severely affected the temperature stability of the desorption point. As a consequence, the product ion ratio of the molecular ion and the dimer ions differed between each sample run, i.e. leading to the less satisfactory RSD.
 | ||
| Fig. 1 Selected ion current (m/z 116) of 6 continuous MIT analyses. The RSD of the peak area is 6.7%. | ||
Fast screening and classifying preservatives in cosmetics with MIPDI-MS
The standard spectra of two preservatives, MIT (Fig. 2(a)) and methyl 4-hydroxybenzoate (M.W. 152, Fig. 2(b)), were obtained, along with their collision induced dissociation (CID) spectra by the MIPDI source. These standard spectra were compared with those obtained from commercial cosmetic samples to further assure confidence in the identification of MIT and methyl 4-hydroxybenzoate in order to avoid false positive tests. The insets of Fig. 2 are the CID spectrum of the corresponding protonated molecular ions. It can be inferred that both MIT (Fig. 2(a), [M + H]+, m/z 116) and methyl 4-hydroxybenzoate (Fig. 2(b), [M + H]+, m/z 153) gave their respective protonated molecular ion. A 25 a.u. collision energy was applied. The featured fragment ions of MIT are at m/z 98.8 and m/z 74.0. For methyl 4-hydroxybenzoate, a 20 a.u. collision energy was applied to obtain the fragment ions. The featured fragment ions of methyl 4-hydroxybenzoate are at m/z 120.8 and m/z 108.8. These feature fragment ions were used for the further proof of the existence of those two substances. | ||
| Fig. 2 Standard spectra of (a) MIT and (b) methyl 4-hydroxybenzoate, the insets are the CID spectra corresponding to the protonated molecular ions. The collision energies are 25 a.u. and 20 a.u. respectively. | ||
Locally purchased sunscreen, facial cream and moisturizer were chosen for the qualitative analysis experiment. The sample preparation was done by smearing the cosmetics onto glass slides. No further pre-treatment was done. The cosmetic samples were desorbed by the MIPDI source directly. The original mass spectra of the samples, corresponding to sunscreen, facial cream and moisturizer, are shown in Fig. 3. What can be found in the spectra is that, despite the complex matrix, the presence of MIT ([M + H]+) is still clearly indicated by the peak at m/z 116. Other peaks in the spectra show the ions of ingredient substances in the cosmetic samples desorbed along with MIT.
 | ||
| Fig. 3 Full scan spectra of commercial cosmetic samples: (a) sunscreen; (b) facial cream; (c) moisturizer. The spectra were obtained from direct desorption ionization. | ||
A deficiency that vexes the users of traditional chromatography is the limited sampling speed when chromatography is performed in the qualitative application of bulk samples. The MIPDI source is advantageous compared with the chromatography method, because the MIPDI source is exactly suitable to instantly and qualitatively say “yes/no” with regard to one or several substances in a sample, i.e. the MIPDI source possesses great potential in fast screening applications. As a very crucial characteristic of the MIPDI source, the fast classification capability of cosmetic samples was examined in this research. In this section, a classification process of commercially available cosmetics according to the added preservatives is described using the MIPDI source. There were 5 commercially available cosmetic samples from a local cosmetic shop used in this experiment. The selected cosmetics possibly contained MIT and methyl 4-hydroxybenzoate. The cosmetic samples applied to glass slides were randomly analyzed. Depicted in Fig. 4(a) is the real time total ion current (TIC) of the 5 individual cosmetic samples analyzed with the MIPDI source. Fig. 4(b) and (c) are the extracted ion currents of m/z 116 (MIT, [M + H]+) and m/z 153 (methyl 4-hydroxybenzoate, [M + H]+) respectively. The time axes of the three chromatograms are parallel. The TIC was indicative of the time over which the samples were desorbed. The extracted ion currents were used to determine if the corresponding ion appeared when the samples were desorbed. Accordingly, it is self-evident that the 5 samples were classified into two groups. Samples 3, 4 and 5 appear to use MIT as a preservative. Samples 1, 2, 3 and 5 appear to contain methyl 4-hydroxybenzoate as a preservative. This was a surprise, since the ingredient lists from the cosmetic packages state that only 3 of the samples contain methyl 4-hydroxybenzoate and that one sample contains both of the preservatives. There must be a false positive result in sample 3 or 5 with respect to the existence of methyl 4-hydroxybenzoate.
 | ||
| Fig. 4 (a) TIC of the fast screening test (no pre-treatment) of 5 commercially available cosmetics analyses by MIPDI. (b) Extracted ion current at m/z 116 (MIT dosage). (c) Extracted ion current at m/z 153 (methyl 4-hydroxybenzoate dosage). | ||
To identify the potential false positive in the measurement, another verification experiment was conducted. A 20 a.u. collision energy was applied to the ion at m/z 153 in samples 3 and 5 to obtain the CID spectra. The obtained CID spectra were compared to the standard CID spectrum (20 a.u. collision energy) of methyl 4-hydroxybenzoate (Fig. 2(b)). Two major fragments at m/z 120.8 and m/z 108.8 for methyl 4-hydroxybenzoate in the standard CID spectrum did not exist in the CID spectrum of sample 5. Therefore, sample 5 was excluded from the methyl 4-hydroxybenzoate containing list.
The whole classification procedure took less than 5 minutes including sample preparations, equal to 1 sample per minute. The classification time can possibly be significantly reduced further if an automatic sampler is used. As an efficient fast screening method, the MIPDI source has proven its usefulness in real sample pre-classification before going through a more quantitative analysis such as HPLC or GC. The pre-classification ability of the MIPDI source made it possible to do a preliminary screening and classification of an unknown sample with one scan.
Quantitative analysis of preservatives in cosmetics
The quantitative analysis ability of MIPDI-MS was explored for the first time in the present investigation through the standard adding method and standard calibration curve method. To simulate real world analysis conditions, both solid state and liquid state commercial cosmetics were used. The target preservative was MIT. The standard adding method was used for the quantitative analysis of liquid samples and the standard calibration curve method was used for the quantitative analysis of solid samples. Each data point was acquired with a set of 6 individual repetitions in the following experiment.When using the standard adding method, the standard substance is required to be uniformly mixed into the sample substrate. Liquid state samples are suitable to use for this method due to the favorable dispersion of the preservative solutions. Consequently, the standard adding method was used to quantify the concentration of MIT in the commercial moisturizer which is a kind of liquid state cosmetic. This method can efficiently eliminate the matrix effect, i.e. matrix suppression or enhancing can be ignored. To prepare the gradient solution, different volumes (10 μL, 20 μL, 40 μL, 50 μL) of a standard MIT solution (59.6 mg mL−1), used as internal standard, were added into 5 mL samples of a 1:1 methanol diluted commercial sample respectively. The difference in the solvent volume was compensated by adding methanol (40 μL, 30 μL, 10 μL, 0 μL respectively). The obtained gradient sample solutions were directly analyzed with the MIPDI source as described. Fig. 5(a) is the quantitative analysis curve (R2 = 0.91). The x-axis represents the volume of added standard MIT solution. The y-axis represents the integration of the ion current intensity. If the curve is extended to reach the x-axis, the absolute value of x on the intersection point is the equivalent volume of the standard MIT (59.6 mg mL−1) in the original sample. Therefore, the weight of the MIT in the cosmetic sample can be calculated. The MIPDI source quantitative analysis result was 0.14% (w/w). It should be noted that the RSD of the result was the average RSD of the points used to establish the quantitative analysis curve. The relative standard deviation was 8.3%.
 | ||
| Fig. 5 (a) Calibration curve of a liquid state commercial cosmetic sample using the standard addition method. The quantitation equation is y = 194x + 12104, R2 = 0.915. (b) Calibration curve of a solid state commercial cosmetic sample extract using the standard calibration curve method. The quantitation equation is y = 2.79x + 1254, R2 = 0.999. The round symbol indicates the concentration used to establish the calibration curve and the square symbol indicates the sample concentration found on the curve with standard deviations. | ||
The above standard adding method is not suitable for the quantitative analysis of solid samples because a different volume of an internal standard was hard to uniformly add into the sample matrix. Consequently, an extraction procedure was demanded. The extracted sample solution is clean with no interference from the sample matrix. After extraction, the sample solution had a simple matrix (methanol as solvent), so the calibration curve method was suitable for use in this situation. The extraction procedure was the same as recommended in the NHSC standard for the preparation of an HPLC sample. The extracted solutions were diluted to 1% (w/w) and analyzed with the MIPDI source. At the same time, a calibration curve was established with the MIPDI source using pure MIT standard solution. Fig. 5(b) is the calibration curve (R2 = 0.99). The circles which were used for establishing the calibration curve represent the MIPDI source results of analyzing the standard solutions. The squares represent the extraction solution signal. To obtain a reliable result, each data point was repeated 6 times. The signal from the extract is shown with the corresponding standard error. The MIPDI-MS quantitative analysis result was 39 ppm (Table 2) and the relative standard deviation of the 6 tests was 5.9%.
| Analysis method | Solid state sample (w/w) | Liquid state sample (w/w) |
|---|---|---|
| Liquid chromatography | 77 ppm ± 0.30% | 0.41% ± 3.7% |
| MIPDI-MS | 39 ppm ± 5.9% | 0.14% ± 8.3% |
| Accuracy | −49% | −66% |
The liquid and solid state cosmetic samples were successfully analyzed quantitatively using the MIPDI source. However, the accuracy of the MIPDI result had to be taken into consideration. A comparison of the quantitative analysis results was made between HPLC and MIPDI-MS (Table 2). The HPLC quantification results were obtained using the method recommended by the NHSC. The results of each quantitative analysis of each of the samples are listed in Table 2. The liquid sample analyzed with MIPDI-MS had an error of −66% when compared to the HPLC result. The solid sample analyzed using MIPDI-MS had a relative error of −49%. Negative deviation was introduced by the MIPDI-MS quantitative analysis process. The accuracy of the quantitative analysis of the MIPDI-MS is not ideal. The deviation of the quantitative analysis data made it difficult to accurately quantify the preservatives in the cosmetics. However, the MIPDI-MS quantitative analysis values were in the same order of magnitude as those of the HPLC method which means that the amount present can be roughly estimated using the MIPDI source i.e. semi-quantitative analysis is feasible using the above two methods. The solid state samples gave better quantitative results by MIPDI-MS because these samples had a much simpler matrix after extraction. For the same reason, the RSD of the solid state sample quantitative analysis is better during analysis.
Quantitative analysis using an isotopically labeled standard
The standard adding method and standard calibration curve method had been proved not to be ideal in accurate quantitative analysis when using MIPDI-MS. The accuracy is acceptable for semi-quantitative analysis (order of magnitude level accuracy). However, the performance should be improved further if the MIPDI method is to be applied for accurate quantitative applications. Moreover, both of the above quantitative analysis methods require complex sample pre-treatment. For the reasons above, those two quantitative analysis methods for MIPDI-MS have no superiority compared with the traditional HPLC method regardless of the operational convenience or quantitative analysis accuracy.As an accurate and fast quantitative method, the isotopic labeled standard28 method has been widely used in HPLC-MS. In a few cases, the isotopic labeled standard method was used in the quantitative analysis31,32 of ambient ionization source mass spectrometry and obtained superior accuracy and reliable results. In this method, the quantitative analysis process does not merely measure the absolute intensity of the analyte ion, but also the relative intensity between the analyte and isotopically labeled standard. In this way, the sampling imprecision and matrix interference can be efficiently reduced. Sampling imprecision is an essential deficiency that strongly affects the quantitative accuracy of the MIPDI source because the desorption process is randomly affected by the surroundings due to the open ion source. The quantitative analysis accuracy will be improved if the isotopic labeled standard method is applied in the quantitative analysis of the MIPDI source. Furthermore, the sample preparation is simplified by using the isotopically labeled method. All that needs to be done is to spike a known concentration of an isotopically labeled standard into the sample. It is necessary to study the application of the isotopically labeled standard method in the MIPDI quantitative analysis.
To demonstrate the accurate quantitative analysis ability of the MIPDI source, a conceptual experiment was conducted using the isotopic labeled standards method. The confirmatory experiment was aimed at validating if the isotopic labeled standard method can be used in the quantitative detection of preservatives in a cosmetic matrix using an MIPDI source. During the experiment, caffeine was regarded as a preservative and was added into cosmetics which were called “matrix matched cosmetic samples”. It was essential to check if the peak of the reference D3-caffeine (m/z 198.0) was affected by the matrix before quantitative analysis, because spectral overlap can lead to negative deviation, so blank “matrix matched cosmetic samples” were analyzed using the MIPDI source. As can be seen in Fig. 6, there is no interference peak at the position of m/z 198.0 in the “matrix matched cosmetic sample”. The average intensity at m/z 198.0 is 0.4 a.u. Such a weak signal intensity cannot affect the signal accuracy of D3-caffeine, i.e. the further quantitation process cannot be affected by spectral overlap.
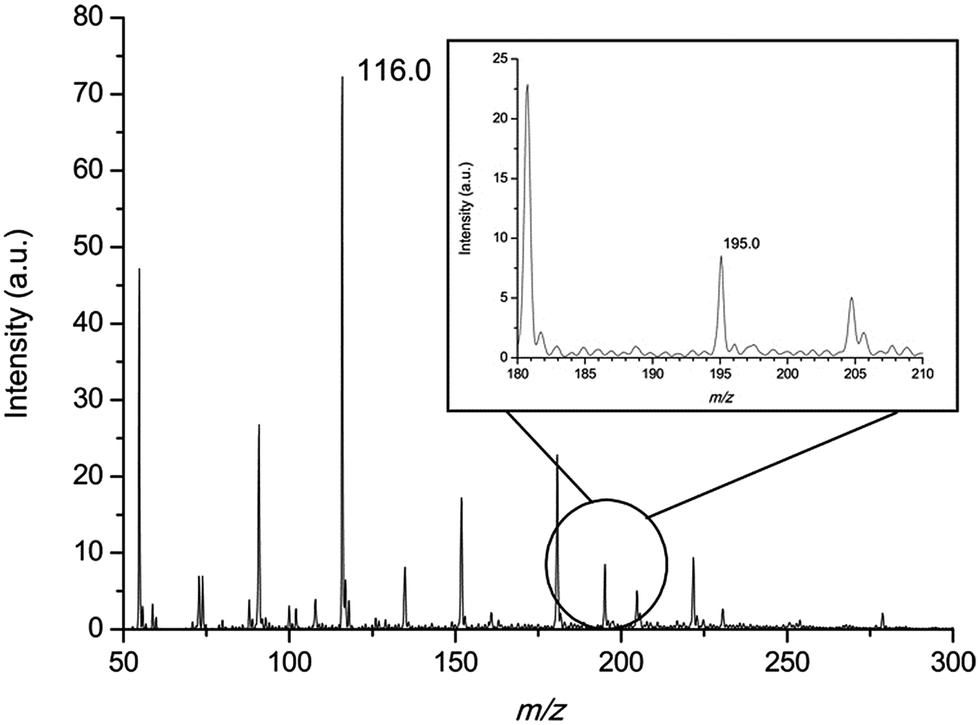 | ||
| Fig. 6 Mass spectrum of “matrix matched cosmetic sample” spiked with caffeine; the inset is the detailed mass spectrum around m/z 195.0 (protonated caffeine). | ||
The “matrix matched cosmetic samples” were prepared as described in the experimental section. To quantify the concentration of the “unknown” concentration of caffeine (125 μg mL−1) in the cosmetic matrices, a comparison was made between the peak height of the caffeine (m/z 195.0, [M + H]+) and that of the spiked isotopic labeled standard D3-caffeine (m/z 198.0, [M + H]+) which are ionized at the same time through the MIPDI source. The peak height ratio equals the concentration ratio, and thus the concentration of caffeine can be calculated. Fig. 7 is one of the MIPDI mass spectra used for quantitative analysis. The peaks of caffeine (m/z 195.0, [M + H]+) and D3-caffeine (m/z 198.0, [M + H]+) can be clearly identified along with the ionized matrices. The main preservative, MIT ([M + H]+), can also be clearly identified at m/z 116.0. The quantitative analysis experiments were performed with 4 different concentrations of reference D3-caffeine, ranging from 21.5 μg mL−1 to 86.0 μg mL−1. Measurement of each concentration was repeated 10 times. The results show that the accuracy of MIPDI quantitative analysis is within −3.9% to +5.1%. The RSD was as low as 5.5%. The detailed information is generalized in Table 3, including the ion intensity ratio, quantitation result, relative standard deviation and accuracy.
 | ||
| Fig. 7 Mass spectrum of a “matrix matched cosmetic sample” spiked with caffeine ([M + H]+, m/z 195.0) and D3-caffeine ([M + H]+, m/z 198.0). | ||
| D3-caffeine label concentration (μg mL−1) | Peak height ratio of D3-caffeine/caffeine | Calculated concentration of caffeine (μg mL−1) | Accuracy |
|---|---|---|---|
| 21.5 | 0.168 ± 5.5% | 128 | +2.4% |
| 43.0 | 0.354 ± 6.0% | 122 | −2.8% |
| 64.5 | 0.536 ± 9.1% | 120 | −3.9% |
| 86.0 | 0.654 ± 8.1% | 131 | +5.1% |
To further minimize the influence of random error, the relative intensity between the caffeine ion and D3-caffeine ion was used to establish a calibration curve. The x-axis is the concentration of D3-caffeine and the y-axis is the peak intensity ratio of D3-caffeine and caffeine (D3-caffeine/caffeine). The spots used to establish the calibration curve are shown with error bars (Fig. 8). The linearity (R2) of the calibration curve is 0.985. The concentration of caffeine in the “matrix matched cosmetic sample” can be calculated through the equation y = 0.0077x + 0.0114. The calculated x is the concentration of caffeine, when y = 1. This means that when the peak intensity ratio of D3-caffeine and caffeine is equal to 1, the concentration of caffeine equals D3-caffeine. In this method, the quantitative analysis result of unknown caffeine is 129 μg mL−1 and the accuracy is +3.2%.
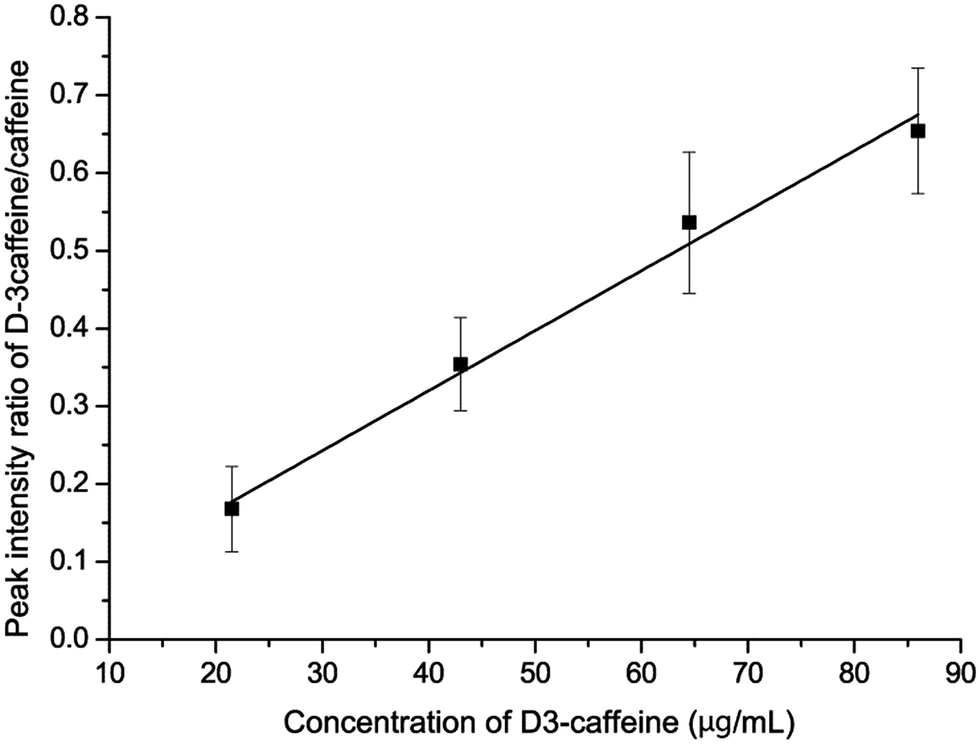 | ||
| Fig. 8 Calibration curve of the isotopically labeled standard method. The curve can be described by the equation y = 0.0077x + 0.0114 (R2 = 0.985). | ||
The above results suggested that the isotopically labeled standard is a potential methodology for the quantitative analysis of cosmetic preservatives using MIPDI-MS. The RSD of the individual test reached 5.5%. The quantitative analysis ability of MIPDI-MS is competitive to those of other ambient ionization sources in such a harsh matrix. The RSD value also approached that of a typical HPLC/GC method (5%), which is satisfactory. The average accuracy of the 4 groups of tests is 3.6%, meaning that the MIPDI can accurately quantify the concentration of cosmetic preservatives using the isotopically labeled standard method. In addition, no sample extraction was done throughout the isotopically labeled standard method quantitative analysis process. All that needed to be done for the sample preparation was to add the isotopically labeled standard.
This is the first report of accurately and quantitatively analyzing trace amounts of samples without sample extractions in complex matrices using MIPDI-MS. This report sets an example of the quantitative analysis by an MIPDI source, meaning that MIPSI-MS can potentially be applied to other fields such as drugs, foods and environmental analysis. Also, MIPDI-MS can be a good alternative to the traditional HPLC/GC method.
Conclusions
The microwave induced plasma desorption ionization (MIPDI) source has demonstrated its worthiness and effectiveness in the fast detection of trace amounts of preservatives in various commercial cosmetics. The quantitative and qualitative analysis behavior of the MIPDI source has been investigated for the first time. The MIPDI source provides as low as a 3.0 pg mm−2 detection limits for 6 commonly used cosmetic preservatives. The relative standard error of individual MIPDI tests was as low as 5.2%. Direct analysis of commercially available cosmetics without any sample pretreatment was also reported, covering solid and liquid state cosmetics. The fast screening capability was investigated by the blind analysis of 5 commercially available cosmetic samples. The cosmetics were successfully classified into two categories according to the added preservatives. Quantifying the preservatives in the cosmetics using the traditional standard adding method and the calibration curve method proved to be less effective in accurately quantifying the preservatives in cosmetics even though sample pretreatment was done. The standard adding method (liquid state cosmetics, without extraction) led to a 66% negative deviation and the calibration curve method (solid state cosmetics, with extraction) led to a 49% negative deviation, which was validated using HPLC. A conceptual experiment was conducted using an isotopically labeled standard. Using the method, the concentration of caffeine in cosmetic matrices was quantified satisfactorily with no sample pretreatment, with an RSD value of 5.5% and accuracy of 3.6%. The approaches established in this work indicate that the MIPDI source is a promising tool for future applications where rapid qualitative and quantitative analysis is needed.Acknowledgements
The authors thank the National Recruitment Program of Global Experts (NRPGE, 0216) and the Hundred Talents Program of Sichuan Province (HTPSP, 036) for financially supporting this research.References
- L. Sanchez-Prado, J. P. Lamas, M. Lores, C. Garcia-Jares and M. Llompart, Anal. Chem., 2010, 82, 9384–9392 CrossRef CAS PubMed.
- C. Selvius DeRoo and R. A. Armitage, Anal. Chem., 2011, 83, 6924–6928 CrossRef CAS PubMed.
- S. Yang, J. Han, Y. Huan, Y. Cui, X. Zhang, H. Chen and H. Gu, Anal. Chem., 2009, 81, 6070–6079 CrossRef CAS.
- I. Cotte-Rodríguez, H. Hernández-Soto, H. Chen and R. G. Cooks, Anal. Chem., 2008, 80, 1512–1519 CrossRef PubMed.
- E. Block, A. J. Dane, S. Thomas and R. B. Cody, J. Agric. Food Chem., 2010, 58, 4617–4625 CrossRef CAS PubMed.
- D. Huang, L. Luo, C. Jiang, J. Han, J. Wang, T. Zhang, J. Jiang, Z. Zhou and H. Chen, J. Agric. Food Chem., 2011, 59, 2148–2156 CrossRef CAS PubMed.
- R. Kubec, R. B. Cody, A. J. Dane, R. A. Musah, J. Schraml, A. Vattekkatte and E. Block, J. Agric. Food Chem., 2010, 58, 1121–1128 CrossRef CAS PubMed.
- H. Zhang, L. Zhu, L. Luo, N. Wang, K. Chingin, X. Guo and H. Chen, J. Agric. Food Chem., 2013, 61, 10691–10698 CrossRef CAS PubMed.
- N. E. Manicke, A. L. Dill, D. R. Ifa and R. G. Cooks, J. Mass Spectrom., 2010, 45, 223–226 CrossRef CAS PubMed.
- J. Hajslova, T. Cajka and L. Vaclavik, TrAC, Trends Anal. Chem., 2011, 30, 204–218 CrossRef CAS PubMed.
- L. Ackerman, G. Noonan and T. Begley, Food Addit. Contam., 2009, 26, 1611–1618 CrossRef CAS PubMed.
- T. J. Kauppila, J. M. Wiseman, R. A. Ketola, T. Kotiaho, R. G. Cooks and R. Kostiainen, Rapid Commun. Mass Spectrom., 2006, 20, 387–392 CrossRef CAS PubMed.
- D. J. Weston, R. Bateman, I. D. Wilson, T. R. Wood and C. S. Creaser, Anal. Chem., 2005, 77, 7572–7580 CrossRef CAS PubMed.
- H. Chen, N. N. Talaty, Z. Takáts and R. G. Cooks, Anal. Chem., 2005, 77, 6915–6927 CrossRef CAS PubMed.
- A. L. Dill, D. R. Ifa, N. E. Manicke, A. B. Costa, J. A. Ramos-Vara, D. W. Knapp and R. G. Cooks, Anal. Chem., 2009, 81, 8758–8764 CrossRef CAS PubMed.
- J. M. Wiseman, D. R. Ifa, Y. Zhu, C. B. Kissinger, N. E. Manicke, P. T. Kissinger and R. G. Cooks, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18120–18125 CrossRef PubMed.
- J. M. Wiseman, S. M. Puolitaival, Z. Takáts, R. G. Cooks and R. M. Caprioli, Angew. Chem., 2005, 117, 7256–7259 CrossRef PubMed.
- X. Ding, X. Zhan, X. Yuan, Z. Zhao and Y. Duan, Anal. Chem., 2013, 85, 9013–9020 CrossRef CAS PubMed.
- T. Zhang, W. Zhou, W. Jin, J. Zhou, E. Handberg, Z. Zhu, H. Chen and Q. Jin, J. Mass Spectrom., 2013, 48, 669–676 CrossRef CAS PubMed.
- X. Zhan, Z. Zhao, X. Yuan, Q. Wang, D. Li, H. Xie, X. Li, M. Zhou and Y. Duan, Anal. Chem., 2013, 85, 4512–4519 CrossRef CAS PubMed.
- Z. Zhao, D. Li, B. Wang, X. Ding, J. Dai, X. Yuan, X. Li and Y. Duan, Int. J. Mass Spectrom., 2015, 376, 65–74 CrossRef CAS PubMed.
- A. Albert and C. Engelhard, Anal. Chem., 2012, 84, 10657–10664 CrossRef CAS PubMed.
- C. Chang, G. Xu, Y. Bai, C. Zhang, X. Li, M. Li, Y. Liu and H. Liu, Anal. Chem., 2012, 85, 170–176 CrossRef PubMed.
- B. Gilbert-López, M. Schilling, N. Ahlmann, A. Michels, H. Hayen, A. Molina-Díaz, J. F. García-Reyes and J. Franzke, Anal. Chem., 2013, 85, 3174–3182 CrossRef PubMed.
- J. M. Nilles, T. R. Connell and H. D. Durst, Anal. Chem., 2009, 81, 6744–6749 CrossRef CAS PubMed.
- S.-Y. Wong and Y.-C. Chen, J. Mass Spectrom., 2014, 49, 432–436 CrossRef CAS PubMed.
- Z. Yang, J. Pavlov and A. B. Attygalle, J. Mass Spectrom., 2012, 47, 845–852 CrossRef CAS PubMed.
- J. F. García-Reyes, A. U. Jackson, A. Molina-Díaz and R. G. Cooks, Anal. Chem., 2008, 81, 820–829 CrossRef PubMed.
- S. Yu, E. Crawford, J. Tice, B. Musselman and J.-T. Wu, Anal. Chem., 2008, 81, 193–202 CrossRef PubMed.
- L. Dai, F. Meng, Z. Pan, Q. Ni and G. Li, Analytical Chemistry, Higher Education Press, Beijing, China, 4 edn, 2000 Search PubMed.
- L. Nyadong, S. Late, M. Green, A. Banga and F. Fernández, J. Am. Soc. Mass Spectrom., 2008, 19, 380–388 CrossRef CAS PubMed.
- N. Talaty, C. C. Mulligan, D. R. Justes, A. U. Jackson, R. J. Noll and R. G. Cooks, Analyst, 2008, 133, 1532–1540 RSC.
| This journal is © The Royal Society of Chemistry 2015 |