
Simultaneous quantification of multiple volatile active components in rat plasma using a headspace-solid phase dynamic extraction method coupled to gas chromatography-tandem mass spectroscopy: application in a pharmacokinetic study of Longhu Rendan pills
Tian-Ming Wanga,
Li-Qing Dingb,
Hua-Jia Jinb,
Rong Shia,
Jia-Sheng Wua,
Li Zhub,
Yi-Qun Jia*c and
Yue-Ming Ma*a
aDepartment of Pharmacology, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China. E-mail: mayueming_117@hotmail.com; Fax: +86 21 5132 2386; Tel: +86 21 5132 2386
bShanghai Zhonghua Pharmaceutical Co., Ltd, Shanghai 200052, China
cExperiment Center for Science and Technology, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China. E-mail: yqjia1961@126.com; Fax: +86 21 5132 2686; Tel: +86 21 5132 3028
First published on 19th March 2015
Abstract
Longhu Rendan pills (LRPs), a traditional Chinese over-the-counter medicine, have been used for the prevention and treatment of heatstroke and motion sickness. A sensitive, specific, and accurate headspace-solid-phase dynamic extraction method coupled to gas chromatography-tandem mass spectrometry (HS-SPDE-GC-MS/MS) was developed and validated for the investigation of the pharmacokinetic properties of L-menthol, borneol, isoborneol, and the metabolite camphor in rats after oral administration of LRPs. Target compounds were extracted using an SPDE needle device coated with a polydimethylsiloxane solid phase. Detection of components was achieved by GC-MS/MS in multiple reaction monitoring mode. This method was successfully applied in the evaluation of the pharmacokinetics of components and a metabolite of LRPs after a single intragastric administration of a 0.92 g kg−1 dose to rats. Pharmacokinetic parameters were calculated from the plasma concentration–time data. Cmax values of L-menthol, borneol, isoborneol, and camphor in rat plasma were determined to be 876 ± 341, 268 ± 149, 158 ± 91, and 126 ± 56 ng mL−1, respectively, and the AUC0–t values were measured as 876 ± 259, 408 ± 121, 140 ± 50, and 401 ± 35 ng h mL−1, respectively. These results provide useful information on the effective components of LRPs.
1. Introduction
Longhu Rendan pills (LRPs), a classic traditional Chinese over-the-counter medicine, are composed of Mentholum, Borneolum Synthcticum, Flos Caryophylli, Fructus Anisi Stellati, Radix Aucklandiae, Fructus Amomi, Cortex Cinnamomi, Fructus Piperis, Rhizoma Zingiberis, Catechu, and Radix Glycyrrhizae. LRPs have been used for more than a century in China and are licensed by the State Food and Drug Administration (SFDA) of China (no. Z20025168). LRPs have been widely used for the prevention and treatment of heatstroke and motion sickness. Modern pharmacological studies have confirmed that LRPs elicit significant anti-heatstroke and anti-motion sickness activity, and exhibit peripheral antiemetic effects in rats.1 However, there is currently no published information regarding the pharmacokinetics of LRPs, which would allow us to understand the pharmacological mechanisms underlying the therapeutic effects of LRPs.LRPs contain a number of volatile compounds that elicit a variety of pharmacological effects. Menthol causes gastric relaxation by reducing acetylcholine release2 and shows antiemetic,3 anti-inflammatory, analgesic,4 and anti-peristaltic properties.5 Borneol and isoborneol exert anti-inflammatory,6 analgesic,6,7 and neuroprotective effects.8,9 Furthermore, borneol inhibits acetylcholine-mediated effects10 and shows anti-coagulant11 and vasorelaxant activities.12 Moreover, borneol can enhance the oral bioavailability and distribution of drugs to the brain tissue as well as penetrate the blood-brain barrier.13,14 Borneol and isoborneol can be oxidized to camphor in mice, rats, and rabbits.15 Camphor has analgesic16 and vasorelaxant activities.17 Moreover, menthol and camphor have been shown to act synergistically.18 Although there is no report on the anti-heatstroke and anti-motion sickness of L-menthol, borneol, isoborneol, and camphor, respectively, it has been reported that the anti-inflammatory, analgesic and neuroprotective, and anti-coagulant and vasorelaxant properties may contribute to anti-heatstroke effects,19 and the anticholinergic, gastric relaxation, antiemetic, anti-inflammatory, analgesic, and anti-peristaltic effects may contribute to the anti-motion sickness.20 Therefore, we hypothesized that L-menthol, borneol, isoborneol, and camphor contribute to the therapeutic efficacy of LRPs, and that they are the major bioactive ingredients in LRPs. In order to improve our understanding of the mechanisms underlying the therapeutic effects of LRPs, it is important to study the pharmacokinetics of L-menthol, borneol, isoborneol, and camphor after the oral administration of LRPs.
Volatile compounds are commonly analysed using gas chromatography-tandem mass spectrometry (GC-MS/MS). Conventional pre-treatment methods such as liquid–liquid extraction (LLE) used for quantifying the concentration of compounds in biological samples can cause significant evaporative losses of the volatile components, which are hard to enrich, resulting in the loss of sensitivity and unacceptable assay accuracy. These factors make the sensitive and accurate quantification of volatile components in biological samples very challenging. Solid-phase dynamic extraction (SPDE) developed by Chromtech (Idstein, Germany) in 2000 is the first commercially available inside-needle device.21 SPDE has the advantages of high sensitivity, short sample preparation and extraction times, and high sample throughput, in part reflecting the full automation of the method. It has been extensively used in environmental, pharmaceutical, and biomedical studies as a solvent-free technique for the extraction, concentration, and desorption of volatile compounds.22–27 To the best of our knowledge, there is only one report published to date describing a pharmacokinetic study using the HS-SPDE-GC-MS/MS approach.28 However, the method described in that publication is not suitable for the analysis of LRPs because of the lower sensitive quantification of borneol and isoborneol and the incapacity to detect L-menthol and camphor in plasma. To address this challenge, we developed and validated an accurate, sensitive, and reliable HS-SPDE-GC-MS/MS method for the simultaneous measurement of the levels of L-menthol, borneol, isoborneol, and the metabolite camphor (Fig. 1) in rat plasma. This method was successfully applied in a pharmacokinetic study of volatile compounds found in LRPs.
 | ||
| Fig. 1 Chemical structures of all the analytes. | ||
2. Experimental
2.1. Chemicals and reagents
Camphor, L-menthol, isoborneol, borneol, and naphthalene (purity > 98%) were purchased from the Chinese Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). LRPs were provided by Shanghai Zhonghua Pharmaceutical Co., Ltd (Shanghai, China). By using gas chromatography coupled with triple quadrupole mass spectrometry,29 the levels of menthol, isoborneol, and borneol in LRPs were determined to be 22.7, 5.7, and 9.7 mg g−1, respectively. Ethyl acetate was obtained from Sinopharm Chemical Reagent Co, Ltd. (Shanghai, China). Ultra-pure water was purified using a Milli-Q system (Millipore, Bedford, MA, USA).2.2. Animals
Male Wistar rats, weighing 250 ± 20 g (grade II, certificate no. SCXK 2012-0002) were purchased from Shanghai SLAC Laboratory Animal Co. Ltd. They were maintained on a 12 h light–dark cycle in an environmentally controlled breeding room (temperature 22–25 °C, humidity 60% ± 5%) for 7 days. The animals were fasted for 12 h prior to the experiments, but continued to have free access to water during this time. All animal experiments were conducted in accordance with the National Research Council guidelines.2.3. Instrumentation and analytical conditions
Analysis was performed using an Agilent 7890A GC interfaced to a Triple Quadrupole Mass Spectrometer Agilent 7000B (Agilent Technologies, California, USA). Data acquisition, processing, and evaluation were performed using Masshunter software, version B.05.02 1032 (Agilent Technologies). Chromatographic separation was performed on a VF-WAXms capillary column (30 m × 0.25 mm ID; Agilent Technologies) coated with 100% polyethylene glycol (0.25 μm film thickness).The following temperature program was used: 50 °C (0 to 1 min), 50 to 150 °C (1 to 9.3 min at 12 °C min−1), 150 to 200 °C (9.3 to 11.8 min at 20 °C min−1), 200 to 245 °C (11.8 to 12.8 min at 45 °C min−1), with the system held at 245 °C for 2 min. Helium and nitrogen were used as collision cell gases at flow rates of 2.25 and 1.5 mL min−1, respectively, with helium used as the carrier gas at a constant flow rate of 2.5 mL min−1. The temperatures of the transfer line and the ion source were set to 250 and 300 °C, respectively. The solvent delay was set to 6 min in splitless mode. The mass detector was operated in electron impact ionisation (EI) MS/MS mode at 70 eV using multiple reaction monitoring (MRM) for quantification of all analytes. The full list of the analytes, with their time segments, respective retention times, detected ions, dwell times, collision energies, and gains, is presented in Table 1.
| Compound | Time segments (min) | RT (min) | Detected ion (m/z) | Dwell (ms) | CE (V) | Gain |
|---|---|---|---|---|---|---|
| Camphor | 6.0 | 8.34 | 95–95 | 100 | 5 | 30 |
| L-Menthol | 6.0 | 9.64 | 95–95 | 100 | 5 | 30 |
| Isoborneol | 6.0 | 9.93 | 95–95 | 100 | 5 | 30 |
| Borneol | 6.0 | 10.27 | 95–95 | 100 | 5 | 30 |
| Naphthalene | 10.5 | 10.69 | 128–102 | 100 | 25 | 30 |
SPDE was performed using a CTC-Combi-PAL autosampler supplied by Chromtech (Idstein, Germany). CTC-Combi-PAL autosampler included a single magnet mixer, a gas station to aspire desorption gas and a heated flushing station for conditioning and reconditioning of the SPDE needles (Chromtech). All SPDE sampling steps were automatically controlled by the CTC-Combi-PAL software. The internal surface of the SPDE needle was coated with a PDMS phase with film thickness of 50 μm and film length of 56 mm.
Aliquots (100 μL) of plasma spiked with 10 μL of internal standard (IS) naphthalene (100 ng mL−1) were placed into 10 mL vials and vortex-mixed for 30 s. Before the measurements were obtained, samples were kept at 85 °C for 5 min in a single magnet mixer to reach equilibrium between the HS compartment and the water phase. Following equilibration, a needle was inserted 20 mm into the sample vial to extract the sample. A desorption volume of 1 mL of nitrogen gas was subsequently aspirated into the syringe at the gas station and was desorbed into the injector at a flow rate of 50 μL s−1. Following desorption, the needle was removed from the injector and flushed with nitrogen for 6 min in the needle flush station at a temperature of 250 °C, to prevent any carryover effects. The parameters that affect the extraction rate, such as the number of extraction cycles, syringe temperature, and pre-incubation time, were optimised to obtain the highest extraction efficiency.
2.4. Standard solutions and quality-control samples
Stock solutions of camphor, L-menthol, isoborneol, and borneol were prepared in ethyl acetate at concentrations of 0.66, 3.4, 2.0, and 2.0 mg mL−1, respectively. A series of mixed working standards at concentrations in the 0.5–400 ng mL−1 range were prepared for each compound by diluting a mixture of stock solutions in ethyl acetate. Three levels of quality control (QC) samples at concentrations of 1, 20, and 320 ng mL−1 were prepared separately for each compound in plasma in the same manner. Additionally, the stock solution of IS naphthalene was diluted to a concentration of 100 ng mL−1 in ethyl acetate. All solutions were stored at 4 °C.2.5. Method validation
The method was validated according to the guidelines of the U.S. Food and Drug Administration (FDA).2.6. Pharmacokinetic study
Blood samples (200 μL) were collected in heparinized 1.5 mL polythene tubes at 0, 0.03, 0.08, 0.25, 0.5, 1, 2, 4, 12, 24, and 48 h after intragastric administration of 0.92 g kg−1 LRPs (equivalent to 20.89 mg kg−1 of L-menthol, 5.25 mg kg−1 of isoborneol, and 8.94 mg kg−1 of borneol)29 to rats. Samples were centrifuged and the isolated plasma was stored at −80 °C until the analysis. Concentrations of analytes were measured in the plasma, as described above. Samples with concentrations above the upper limit of quantification were diluted with blank plasma and re-analysed. The plasma pharmacokinetic parameters were estimated using the non-compartmental model in the WinNonlin software package (Build 6.1.0.173, Pharsight Corporation, MO, USA).3. Results and discussion
3.1. Method development
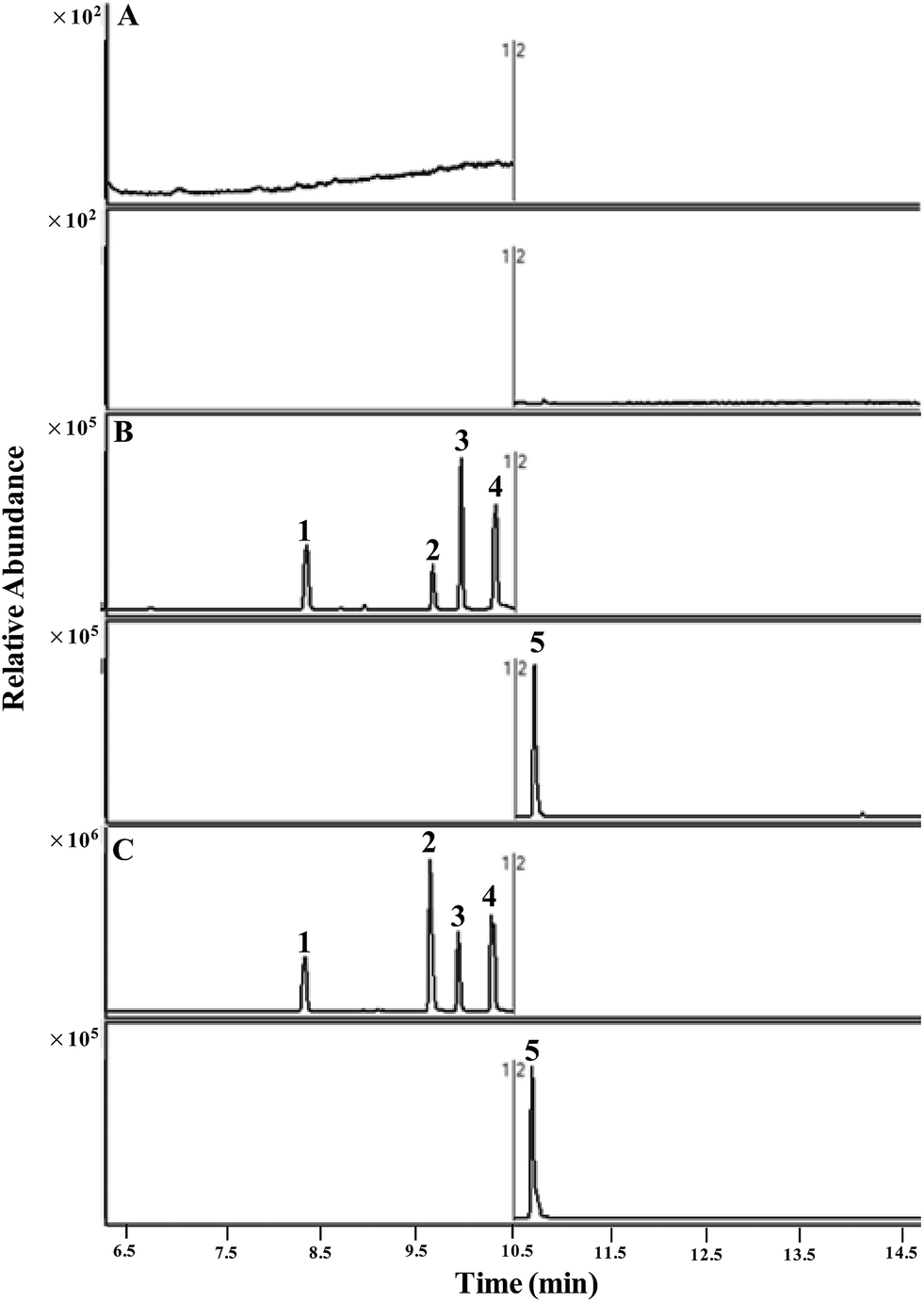 | ||
| Fig. 2 MRM extracted ion chromatograms of (1) camphor, (2) L-menthol, (3) isoborneol, (4) borneol, (5) naphthalene. (A) Blank rat plasma, (B) blank plasma spiked with reference compounds (80 ng mL−1), and (C) plasma sample 30 min after oral administration of LRPs in rats. | ||
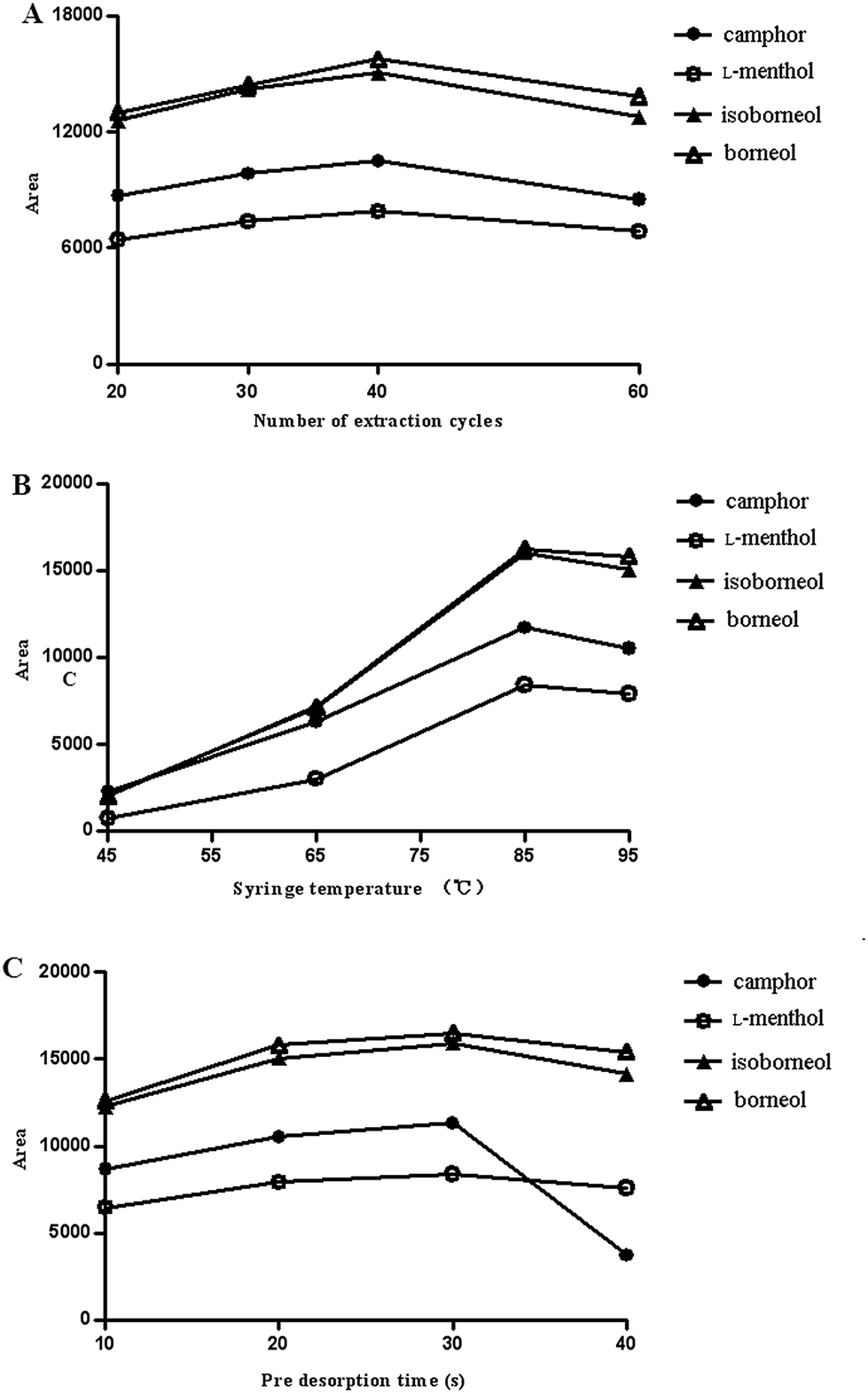 | ||
| Fig. 3 Effect of the extraction parameters on the SPDE efficiency (the concentration of each compound was 30 ng mL−1): (A) number of extraction cycles, (B) syringe temperature and (C) pre desorption time. | ||
3.2. Method validation
| Compounds | Calibration curve | r | Linear range (ng mL−1) | LLOQ (ng mL−1) |
|---|---|---|---|---|
| Camphor | Y = 1.146498X + 0.004245 | 0.9963 | 0.50–400.00 | 0.50 |
| L-Menthol | Y = 0.615042X + 0.002673 | 0.9961 | 0.50–400.00 | 0.50 |
| Isoborneol | Y = 1.612448X + 0.002094 | 0.9963 | 0.50–400.00 | 0.50 |
| Borneol | Y = 1.745362X + 0.014426 | 0.9961 | 0.50–400.00 | 0.50 |
| Compounds | Concentration (ng mL−1) | Intra-day (n = 6) | Inter-day (n = 5) | ||||
|---|---|---|---|---|---|---|---|
| Mean | RSD (%) | Accuracy (%) | Mean | RSD (%) | Accuracy (%) | ||
| Camphor | 1.00 | 1.06 ± 0.08 | 7.07 | 106.20 | 1.04 ± 0.06 | 6.18 | 104.45 |
| 20.00 | 19.04 ± 0.81 | 4.27 | 95.20 | 19.24 ± 1.53 | 7.93 | 96.20 | |
| 320.00 | 321.28 ± 23.83 | 7.42 | 100.40 | 324.94 ± 18.30 | 5.63 | 101.54 | |
| L-Menthol | 1.00 | 0.97 ± 0.06 | 6.38 | 96.65 | 1.01 ± 0.07 | 7.24 | 100.70 |
| 20.00 | 20.39 ± 1.25 | 6.12 | 101.95 | 19.56 ± 1.62 | 8.30 | 97.80 | |
| 320.00 | 315.21 ± 27.76 | 8.81 | 98.50 | 324.00 ± 24.12 | 7.44 | 101.25 | |
| Isoborneol | 1.00 | 1.03 ± 0.05 | 5.33 | 102.90 | 0.99 ± 0.09 | 9.22 | 99.31 |
| 20.00 | 19.55 ± 1.29 | 6.62 | 97.74 | 19.71 ± 1.88 | 9.53 | 98.57 | |
| 320.00 | 312.65 ± 24.68 | 7.90 | 97.70 | 322.15 ± 21.02 | 6.53 | 100.67 | |
| Borneol | 1.00 | 1.05 ± 0.07 | 6.77 | 104.65 | 1.00 ± 0.08 | 7.84 | 100.29 |
| 20.00 | 19.49 ± 1.19 | 6.08 | 97.46 | 19.84 ± 1.52 | 7.68 | 99.19 | |
| 320.00 | 308.21 ± 24.96 | 8.10 | 96.32 | 320.27 ± 24.14 | 7.54 | 100.09 | |
| Compounds | Nominal concentration (ng mL−1) | Autosampler for 12 h stability (%) | Three freeze/thaw cycles at −80 °C stability (%) | Freezing at −80 °C for 15 days stability (%) | Recovery (%) |
|---|---|---|---|---|---|
| Camphor | 1.00 | 98.39 ± 3.93 | 101.90 ± 11.10 | 98.88 ± 4.78 | 88.55 ± 5.16 |
| 20.00 | 92.50 ± 7.20 | 102.61 ± 2.12 | 97.72 ± 0.52 | 83.48 ± 5.62 | |
| 320.00 | 95.94 ± 2.35 | 93.83 ± 5.73 | 105.59 ± 8.24 | 84.71 ± 3.52 | |
| L-Menthol | 1.00 | 98.47 ± 8.93 | 106.86 ± 2.15 | 107.60 ± 3.60 | 78.42 ± 6.48 |
| 20.00 | 93.28 ± 7.01 | 102.29 ± 8.17 | 94.46 ± 4.80 | 74.95 ± 8.23 | |
| 320.00 | 97.43 ± 2.63 | 95.07 ± 5.17 | 108.46 ± 3.52 | 85.40 ± 11.81 | |
| Isoborneol | 1.00 | 92.59 ± 3.19 | 91.65 ± 5.80 | 100.51 ± 3.16 | 79.73 ± 5.64 |
| 20.00 | 93.16 ± 7.50 | 105.63 ± 9.57 | 98.99 ± 3.45 | 79.27 ± 8.00 | |
| 320.00 | 97.96 ± 3.79 | 92.05 ± 6.52 | 109.30 ± 6.12 | 83.00 ± 8.46 | |
| Borneol | 1.00 | 104.92 ± 9.70 | 99.65 ± 5.91 | 104.37 ± 3.93 | 82.52 ± 5.82 |
| 20.00 | 98.78 ± 10.33 | 107.59 ± 7.91 | 93.86 ± 2.20 | 79.27 ± 10.11 | |
| 320.00 | 101.11 ± 1.18 | 95.31 ± 6.13 | 107.86 ± 3.12 | 88.49 ± 8.48 |
3.3. Method applicability
In our present study, the proposed HS-SPDE-GC-MS/MS method for simultaneous quantification of concentrations of camphor, L-menthol, borneol, and isoborneol in rat plasma met the requirements for use in the quantitation of biological samples.Some agents that are commonly used in traditional Chinese medicine, including LRPs, contain multiple volatile ingredients that elicit important pharmacological effects. However, their pharmacokinetics under the common dose have often been unsatisfactorily elucidated to date, mostly due to the shortcomings of conventional pre-treatment methods of biological samples resulting to lower sensitivity of quantification. In our current study, the sensitivity of our proposed method using SPDE coupled to GC-MS for L-menthol, borneol, isoborneol and camphor was 30–100 times higher than that for camphor,31 L-menthol,32 borneol and isoborneol33 using conventional LLE coupled to GC-MS, respectively. Addition to, compared with the reported the method using HS-SPDE-GC-MS/MS approach,26 the present method not only detected borneol and isoborneol with over 40 times higher sensitivity, but also exhibited sufficient sensitivity to determine the levels of L-menthol and camphor in rat plasma. Further, compared with method using LLE in concert with programmable temperature vaporizing-based large-volume injection of the organic extract,34 the present method not only similar sensitively detected borneol, isoborneol, and camphor, but also sensitively determined the levels of L-menthol in rat plasma. The established method was successfully applied in the evaluation of the pharmacokinetics of camphor, L-menthol, borneol, and isoborneol of LRPs after intragastric administration.
Since L-menthol and borneol are aromatic ingredients that are commonly used in many Chinese combination herbal therapies, the method optimized and validated in our current study can also be used in pharmacokinetic studies evaluating related volatile compounds in plasma, following administration of other traditional Chinese medicine agents.
3.4. Pharmacokinetic study
LRPs have been broadly used in China for treatment and prevention of heatstroke and motion sickness, and as an antiemetic agent.1 Despite their widespread use, the pharmacokinetics of LRPs has not yet been investigated. The present study we clarified the pharmacokinetics of camphor, L-menthol, borneol, and isoborneol, after oral administration of LRPs in rats. The concentrations of all ingredients were detectable in rat plasma up to 48 h following oral administration. Fig. 4 shows the mean plasma concentration–time profiles of the investigated components. Calculated pharmacokinetic parameters are presented in Table 5. After oral administration of LRPs, L-menthol, isoborneol, and borneol were rapidly absorbed, with a Tmax value of 0.22 h. Isoborneol and borneol were quickly metabolized to camphor, as evidenced by the fact that the Tmax value of camphor follows closely to those of isoborneol and borneol. All volatile compounds exhibited a half-life of medium length (11–18 h). The bioavailability of borneol and isoborneol determined by calculating the ratio of oral AUC to intravenous AUC was 12.7% and 8.7% in a rat pharmacokinetic study of borneolum.34 In another previous study, the bioavailability of L-menthol was estimated to be about 21% on the basis of the ratio of the 24 h urine excretion of L-menthol glucuronide to the dose35 based on almost all the L-menthol was metabolized into menthol glucuronide and the plasma AUC of menthol glucuronide exceeded 99.5% of the sum of the plasma AUC of L-menthol and the AUC of menthol glucuronide.32 According to these bioavailabilities, the distribution volumes of isoborneol, borneol, and L-menthol were calculated following oral administration of LRPs in our study. The results showed relatively large distribution volumes. Moreover, borneol has been reported to be capable of permeating the blood-brain barrier to reach the brain tissue and the concentration of borneol in the brain is higher than that in serum.36 Taken together, these results suggest that isoborneol, borneol, and L-menthol can be easily distributed into various tissues, including the brain. The study of the pharmacokinetics of volatile compounds from LRPs in our present study provides valuable reference data that can be used to guide the future development of LRPs for clinical use.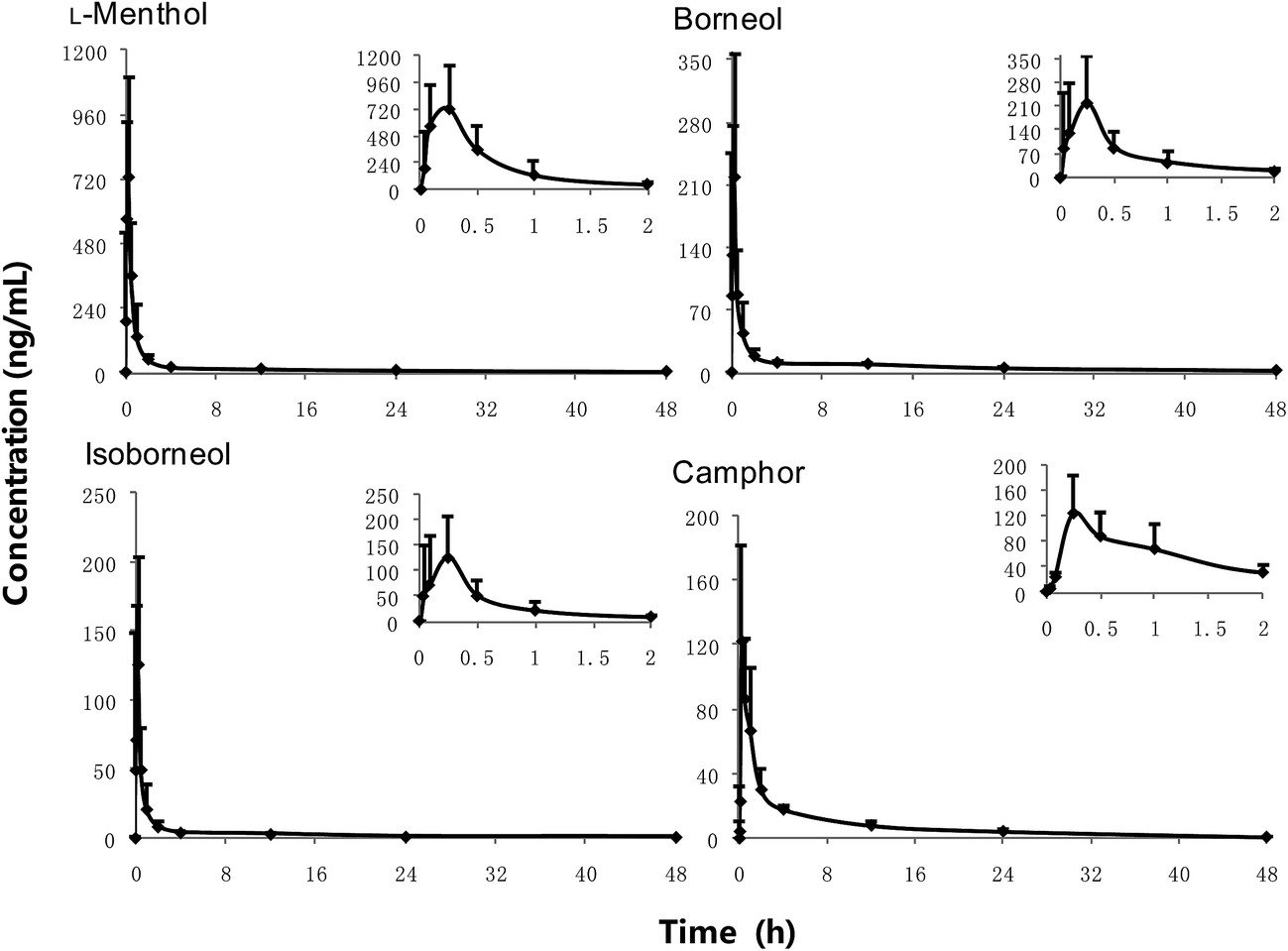 | ||
| Fig. 4 Profiles of mean concentration–time of, L-menthol, borneol, isoborneol and camphor after oral dose of 0.92 g kg−1 Longhu Rendan pills in rats (n = 6, mean ± SD). | ||
| Parameters | L-Menthol | Borneol | Isoborneol | Camphor |
|---|---|---|---|---|
| a -: cannot be calculated. | ||||
| AUC0–t (ng h mL−1) | 876.15 ± 259.22 | 408.19 ± 120.69 | 139.87 ± 49.57 | 401.00 ± 35.07 |
| T1/2 (h) | 16.51 ± 5.73 | 17.56 ± 4.10 | 12.68 ± 4.79 | 11.34 ± 1.71 |
| MRT0–t (h) | 7.34 ± 2.34 | 11.08 ± 2.80 | 6.19 ± 2.64 | 8.95 ± 2.84 |
| Tmax (h) | 0.22 ± 0.07 | 0.22 ± 0.07 | 0.22 ± 0.07 | 0.29 ± 0.10 |
| Cl (L kg−1 h−1) | 4.78 ± 1.11 | 2.56 ± 0.77 | 3.32 ± 1.11 | — |
| Vd (L kg−1) | 113.46 ± 38.94 | 61.82 ± 11.93 | 56.11 ± 15.03 | — |
| Cmax (ng mL−1) | 876.29 ± 341.21 | 267.58 ± 148.82 | 158.07 ± 91.16 | 125.74 ± 55.63 |
Prior to this investigation, to the best of our knowledge, there has been no information on the pharmacokinetics of the bioactive compounds after the oral administration of LRPs, although several pharmacokinetic studies of borneol and isoborneol after intravenous and oral administration33,34,37 and of L-menthol after oral administration32,35 have been reported. In the present study, the elucidation of the pharmacokinetics of L-menthol, isoborneol, borneol, and metabolite camphor following the oral administration of LRPs in rats provides useful information on the bioactive components of LRPs because menthol can reduce acetylcholine release from enteric nerves,2 and borneol inhibits acetylcholine-mediated effects,10 given that anticholinergic effects can help alleviate motion sickness.
In present study, the pharmacokinetic characteristics of volatile compounds from LRPs was only clarified, the pharmacokinetic characteristics of the non-volatile compounds call for further study.
4. Conclusion
A sensitive, specific, accurate, and validated HS-SPDE-GC-MS/MS method was developed for the simultaneous quantification of the levels of L-menthol, isoborneol, borneol, and camphor in rat plasma. The main advantages of this method are its solvent-free nature, high sensitivity, and the technically simple procedure used for plasma sample preparation, based on the HS-SPDE technique. The method was successfully applied in a study evaluating the pharmacokinetics of multiple volatile compounds following oral administration of LRPs.Acknowledgements
The project was supported by Program for Shanghai Innovative Research Team in University (2009), the Shanghai Municipal Education Commission (12QY12 and 2013JW10) and “085” First-Class Discipline Construction of Science and Technology Innovation (085ZY1205).References
- X. H. Li, W. Pan, J. H. Jin, J. L. Du, J. Li and D. F. Song, Pharmacol. Clin. Chin. Mater. Med., 2009, 25, 61–62 Search PubMed.
- A. Amato, R. Serio and F. Mule, Eur. J. Pharmacol., 2014, 745, 129–134 CrossRef CAS PubMed.
- K. Heimes, F. Hauk and E. J. Verspohl, Phytother. Res., 2011, 25, 702–708 CAS.
- C. Y. Liang, W. L. Li, H. Q. Zhang and B. R. Ren, Chin. Wild Plant Resour., 2003, 22, 9–12 Search PubMed.
- A. L. Rozza, C. A. Hiruma-Lima, R. K. Takahira, C. R. Padovani and C. H. Pellizzon, Chem.–Biol. Interact., 2013, 206, 272–278 CrossRef CAS PubMed.
- X. P. Sun, L. J. Ou, S. Q. Mi and N. S. Wang, Tradit. Chin. Drug Res. Clin. Pharmacol., 2007, 18, 353–355 CAS.
- R. E. Granger, E. L. Campbell and G. A. R. Johnston, Biochem. Pharmacol., 2005, 69, 1101–1111 CrossRef CAS PubMed.
- L. L. Tian, Z. Zhou, Q. Zhang, Y. N. Sun, C. R. Li, C. H. Cheng, Z. Y. Zhong and S. Q. Wang, Cell. Physiol. Biochem., 2007, 20, 1019–1032 CrossRef CAS PubMed.
- R. Liu, L. Zhang, X. Lan, L. Li, T. T. Zhang, J. H. Sun and G. H. Du, Neuroscience, 2011, 176, 408–419 CrossRef CAS PubMed.
- T. J. Park, Y. S. Park, T. G. Lee, H. Ha and K. T. Kim, Biochem. Pharmacol., 2003, 65, 83–90 CrossRef CAS.
- Y. H. Li, X. P. Sun, Y. Q. Zhang and N. S. Wang, Am. J. Chin. Med., 2008, 36, 719–727 CrossRef CAS PubMed.
- J. C. Silva-Filho, N. N. P. M. Oliveira, D. D. R. Arcanjo, L. J. Quintans, S. C. H. Cavalcanti, M. R. V. Santos, R. D. C. M. Oliveira and A. P. Oliveira, Basic Clin. Pharmacol. Toxicol., 2012, 110, 171–177 CrossRef CAS PubMed.
- B. Yu, M. Ruan, Y. Sun, X. B. Cui, Y. Yu, L. L. Wang and T. H. Fang, Neural Regener. Res., 2011, 6, 1876–1882 CAS.
- Z. Cai, S. X. Hou, Y. B. Li, B. B. Zhao, Z. X. Yang, S. G. Xu and J. X. Pu, J. Drug Targeting, 2008, 16, 178–184 CrossRef CAS PubMed.
- X. F. Jiang, J. L. Zou, Y. M. Yuan and M. C. Yao, Mode. Tradit. Chin. Med. Mater. Med., 2008, 10, 27–36 Search PubMed.
- H. Xu, N. T. Blair and D. E. Clapham, Journal of Neuroscience, 2005, 25, 8924–8937 CrossRef CAS PubMed.
- A. G. Smith and G. Margolis, Am. J. Pathol., 1954, 30, 857–869 CAS.
- B. L. Xu, Z. R. Wang, K. He and H. X. Chen, Chin. J. Mod. Appl. Pharm., 1998, 15, 32–34 Search PubMed.
- G. S. Lipman, K. P. Eifling, M. A. Ellis, F. G. Gaudio, E. M. Otten and C. K. Grissom, Wilderness Environ. Med., 2014, 25, S55–S65 CrossRef PubMed.
- J. F. Golding and M. A. Gresty, Curr. Opin. Neurol., 2015, 28, 83–88 CrossRef CAS PubMed.
- F. Musshoff, D. W. Lachenmeier, L. Kroener and B. Madea, J. Chromatogr. A, 2002, 958, 231–238 CrossRef CAS.
- C. Bicchi, C. Cordero, E. Liberto, P. Rubiolo and B. Sgorbini, J. Chromatogr. A, 2004, 1024, 217–226 CrossRef CAS PubMed.
- M. A. Jochmann, M. P. Kmiecik and T. C. Schmidt, J. Chromatogr. A, 2006, 1115, 208–216 CrossRef CAS PubMed.
- T. E. Goodwin, M. S. Eggert, S. J. House, M. E. Weddell, B. A. Schulte and L. E. Rasmussen, J. Chem. Ecol., 2006, 32, 1849–1853 CrossRef CAS PubMed.
- M. A. Jochmann, X. Yuan and T. C. Schmidt, Anal. Bioanal. Chem., 2007, 387, 2163–2174 CrossRef CAS PubMed.
- D. Lenz, L. Kroner and M. A. Rothschild, J. Chromatogr. A, 2009, 1216, 4090–4096 CrossRef CAS PubMed.
- B. Rossbach, P. Kegel and S. Letzel, Toxicol. Lett., 2012, 210, 232–239 CrossRef CAS PubMed.
- W. Chang, L. Han, H. Huang, B. Wen, C. Peng, C. Lv, W. Zhang and R. Liu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 963, 47–53 CrossRef CAS PubMed.
- T. M. Wang, L. Q. Ding, Y. Q. Jia, H. J. Jin, R. Shi, L. Zhu and Y. M. Ma, Anal. Methods, 2014, 6, 3713–3719 RSC.
- Y. Li, X. L. Liu, Z. G. Cai and S. X. Zhang, Biomed. Chromatogr., 2014, 28, 193–196 CrossRef CAS PubMed.
- X.-M. Sun, Q.-F. Liao, Y.-T. Zhou, X.-J. Deng and Z.-Y. Xie, J. Pharm. Anal., 2014, 4, 345–350 CrossRef PubMed.
- N. Hiki, M. Kaminishi, T. Hasunuma, M. Nakamura, S. Nomura, N. Yahagi, H. Tajiri and H. Suzuki, Clin. Pharmacol. Ther., 2011, 90, 221–228 CrossRef CAS PubMed.
- T. L. Huang, S. M. Ye, W. P. Ou and S. Q. Mi, Tradit. Chin. Drug Res. Clin. Pharmacol., 2006, 17, 265–267 CAS.
- C. Cheng, X. W. Liu, F. F. Du, M. J. Li, F. Xu, F. Q. Wang, Y. Liu, C. Li and Y. Sun, Acta Pharmacol. Sin., 2013, 34, 1337–1348 CrossRef CAS PubMed.
- A. Gelal, P. Jacob 3rd, L. Yu and N. L. Benowitz, Clin. Pharmacol. Ther., 1999, 66, 128–135 CrossRef CAS PubMed.
- M. R. Liang, Q. D. Liu, T. L. Huang, Y. Q. Zhang and W. P. Ou, Tradit. Chin. Drug Res. Clin. Pharmacol., 1993, 4, 38–40 Search PubMed.
- X. Xu, Y. Li, J. Hou, S. Zhang, Y. Xu, Y. Wang, Y. Zhang, C. Liu and X. He, Planta Med., 2011, 77, 1600–1604 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |