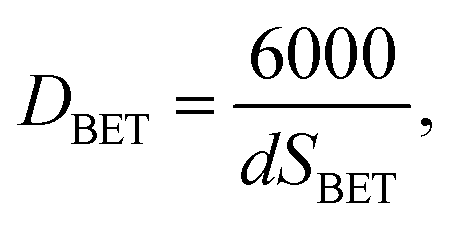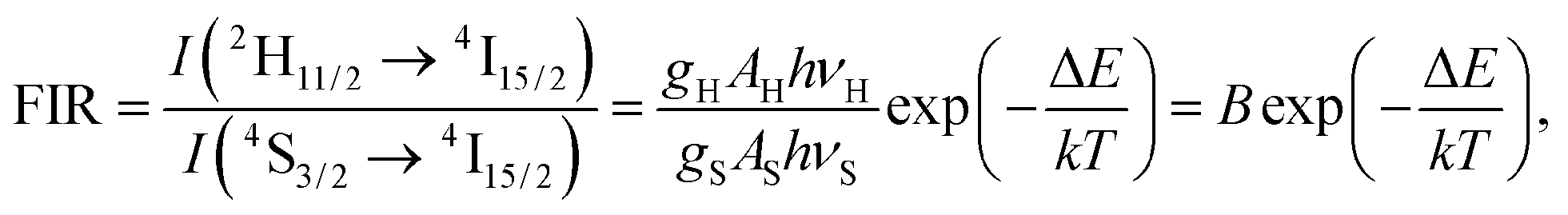DOI:
10.1039/C5RA00675A
(Paper)
RSC Adv., 2015,
5, 27610-27622
An up-converting HAP@β-TCP nanocomposite activated with Er3+/Yb3+ ion pairs for bio-related applications
Received
12th January 2015
, Accepted 26th February 2015
First published on 26th February 2015
Abstract
A HAP@β-TCP nanocomposite doped with Er3+/Yb3+ ion pairs was prepared using Pechini’s technique. The structural properties and morphology of the particles were studied by means of XRD, TEM, and DLS techniques. The cytotoxicity of the developed product was tested on canine osteosarcoma (D17) and murine macrophage (J774.E) cells. Determination of metronidazole release from the HAP@β-TCP nanocomposite was carried out using dynamic dialysis and ultracentrifugation techniques. Thorough analysis of the up-conversion properties of the prepared system was carried out, showing that the GRR depends strongly on the sample temperature induced by the optical density of excitation, and the particle size. Relatively short decay times and the behaviour of the GRR pointed towards the enhanced contribution of non-radiative processes feeding the red emission.
1. Introduction
In recent years nanomaterials and nanocomposites have attracted wide interest due to their unusual mechanical, optical as well as magnetic properties.1,2 Calcium phosphate compounds and their derivatives have been studied for biomedical applications because of their analogy to the inorganic components of organisms.3,4 Phosphate materials with a Ca/P ratio of ∼1.67 form different stable phases such as hydroxyapatite, Ca10(PO4)6(OH)2 (HAP), and other phases such as tricalcium phosphate, Ca3(PO4)2 (α and β-TCP). It is well known that HAP is bioactive and biocompatible with animal tissues, while the others are highly bio-resorbable.5 Furthermore, these compounds display numerous advantages such as bioactivity and biocompatibility that may be favorable for designing fluorescence bioprobes for bio-imaging and bio-sensing.6,7 Although the biomimetic properties of HAP have been intensively studied for bone replacement and drug delivery, not much is known about the possibility of doping these materials with luminescent ions.8,9 In order to control the speed of biodegradation, a biphasic calcium phosphate (BCP) bioceramic containing both HAP and β-TCP is of significant interest.10 The bioactivity and biodegradation of a bioceramic containing both phases depends strongly on the variation of the ratio of HAP/β-TCP. The main difference is that the β-TCP phase is more soluble and undergoes faster resorption than HAP, allowing the precipitation of apatites. In other words osteoconductivity is comparable between HAP and β-TCP.
The spectral properties of apatites doped with rare earth cations such as Nd3+, Yb3+ or Er3+ ions have been studied.11–13 However, these investigations were focused mostly on polycrystalline powders and single crystals, considering their applications as luminescent lamp phosphors and laser hosts. Due to the local structural probing abilities of Eu3+ combined with the presence of hypersensitive transitions, Eu3+ doped apatites have been studied most frequently.14,15 Most of the bioapplications require nanosized materials, but the number of current studies on luminescent HAP nanoparticles is very limited. Highly efficient luminescent nanoparticles are attractive, especially in the field of fluorescence imaging (FI). The most effective are the particles that are able to absorb in the NIR spectral region, because most tissues generate little NIR fluorescence due to the weak NIR absorption, and thus an increase in laser power does not cause any significant damage, in contrast to high-energy UV excitation. Additionally, NIR has a deeper penetration depth than UV or VIS wavelengths and thus is not limited to only shallow tissue imaging.16 One of the promising alternatives lays in the application of inorganic compounds, such as mixed metal oxides or fluorides mutually co-doped with optically active rare earth metals such as Yb3+/Er3+ or Yb3+/Tm3+, showing strong up-conversion.17 The mechanism of this well-known process relies on conversion of the incident infrared light to short wavelength emissions in the visible range. The Yb3+ ions are considered as sensitizers that absorb the pumped light and then transfer the absorbed energy directly to the activators, such as Tm3+ or Er3+. For the up-conversion process it is essential that the energy levels of the sensitizer and activator are in resonance, and this condition assures effective, efficient energy transfer and up-conversion fluorescence.
In the present study we demonstrate thorough studies of the HAP@β-TCP nanocomposite doped with up-converting Er3+/Yb3+ ions, focusing on nanoparticle cytotoxicity and anti-Stokes emission. A few possible implementations of the prepared system in bio-related applications are discussed.
2. Experimental
2.1. Instruments
The development of the crystalline phase was followed by means of XRD, by collecting patterns in the 2θ range of 5–120° with an X’Pert PRO X-ray diffractometer (Cu Kα1 = 1.54060 Å) (PANalytical). The microstructure and morphology of the nanoparticles were investigated by high resolution transmission electron microscopy (HRTEM) using a Philips CM-20 Super Twin microscope, operated at 200 kV. Samples for measurements were prepared by making dispersions of the powders in methanol. Afterwards a droplet of the suspension was deposited on a copper microscope grid covered with perforated carbon. The primary size of the particles was evaluated using a volume weighted formula:| |
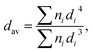 | (1) |
where dav is the average particle size, n is the number of particles and d represents particle diameter.
The BET specific surface area (SBET) was measured by nitrogen gas sorption at 77 K on Quantachrome Autosorb IQ apparatus. Samples were degassed for 18 h at 100 °C before starting. The particle size was calculated from BET measurements using the following equation:
| |
 | (2) |
where
DBET denotes an average particle size (nm),
SBET is the specific surface area (m
2 g
−1) and
d is the density of the investigated material (g cm
−3). The hydrodynamic size was measured using the dynamic light scattering technique with a Nanosight NS 500 automated instrument equipped with a 405 nm laser diode as a light source, backscattered on the nanoparticles. The samples for hydrodynamic size measurements were prepared by taking 1 ml of a water suspension containing the nanoparticles, further diluting this with 19 ml of de-ionized water and transferring it by peristaltic pump to the sample chamber. Typically the starting concentration of nanoparticles in all prepared suspensions was around 1 mg ml
−1. Each measurement was repeated at least three times and conducted with different dilutions of particles to achieve satisfactory statistics and exclude errors connected with too high or too low a number of analyzed objects. From simultaneous measurement of the mean squared displacement of each particle tracked, the particle diffusion coefficient (
Dt) and hence the sphere-equivalent hydrodynamic radius (
rh) can be determined using the Stokes–Einstein equation:
| |
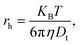 | (3) |
where
KB is Boltzmann’s constant,
T is temperature and
η is solvent viscosity (H
2O). The analysis was done using Nanosight NTA 2.3 software allowing for the determination of particle/object concentration, represented as the number of particles/objects per ml. The up-conversion luminescence spectra were recorded using a Jobin Yvon THR 1000 monochromator equipped with a Hamamatsu R928 photomultiplier and a 1200 grooves per mm holographic grating. As an excitation source, a continuous 975 nm 1.5 W laser diode was used. The luminescence decay times were measured using a LeCroy Wave Surfer oscilloscope using a pulsed 975 nm 10 mJ Ti:sapphire laser pumped by the 532 nm emission of the YAG:Nd
3+ laser. The power dependence of the up-conversion emission intensity
IUPC versus the excitation optical power
Iin of the 975 nm laser diode (CNI laser, China), was measured with a miniature fiber spectrometer (Avantes, Netherlands, spectral resolution ∼3 nm) and fitted with an allometric dependence:
to estimate the order of the up-conversion process, with
N showing the number of photons required to obtain anti-Stokes emission.
2.2. Synthesis of the Ca10(PO4)6(OH)2@β-Ca3(PO4)2:Er3+/Yb3+ nanocomposite
The 2 mol% Er3+ and 10 mol% Yb3+ co-doped HAP@β-TCP nanocomposite powders were prepared by using 2.078 g (8.8 mmol) of Ca(NO3)2·4H2O (99.995% Alfa Aesar), 0.03852 g (0.1 mmol) of Er2O3 (99.99% Alfa Aesar), 0.1970 g (0.5 mmol) of Yb2O3 (99.99% Alfa Aesar) and 0.7923 g (6 mmol) of (NH4)2HPO4 (99.99% Alfa Aesar), using a modified Pechini’s technique. The concentrations of the rare earth cations (RE3+) were set with respect to the overall molar content of Ca2+ cations. Stoichiometric amounts of RE3+ oxides were initially digested in an excess of HNO3 (ultrapure Avantor) in order to transform them into water soluble nitrates, and eventually purified by triple recrystallization. Subsequently, calcium nitrate was dissolved in deionized water and mixed with erbium(III) and ytterbium(III) nitrates. Afterwards 24 g (0.125 mol) of citric acid (99.5% Sigma Aldrich) and 3.4 g (54.78 mmol) of ethylene glycol (ultrapure Avantor) were added under constant stirring at 60 °C, resulting in a viscous mixture. Finally, (NH4)2HPO4 was added and the pH of the solution containing all the substrates was equilibrated with NH3(aq.) (ultrapure Avantor) to achieve a neutral pH. In fact, this step was repeated with additional pH settings (acidic and basic) in order to study the pH effect on the final composition of the product. A turbid solution was obtained as a result of precipitation of by-products (amorphous phosphates). The mixture was further dried for 5 days at 90 °C. The last step of the synthesis involved heat treatment in the temperature range of 800–1000 °C for 3 h. The composition of the obtained samples was determined using inductively coupled plasma atomic emission spectrometry analysis (ICP-AES).
2.3. Determination of metronidazole release from the HAP@β-TCP nanocomposite
Metronidazole (MTZ) loading and release from the HAP@β-TCP nanocomposite (HAP@β-TCP_MTZ) was determined by means of high performance liquid chromatography (HPLC) with UV detection. To determine the total drug loading, 20 mg of powder was dissolved in 20 ml of 4% acetic acid and the resulting solution was analyzed for MTZ concentration. A calibration curve was prepared by spiking a 4% acetic acid water solution with different concentrations of MTZ. The mean loading value (expressed in μg MTZ per mg of HAP@β-TCP) was calculated based on three independent experiments. Drug release was determined by the dynamic dialysis method and ultracentrifugation.
Dynamic dialysis. 20 mg of powder was suspended in 5 ml of water, briefly vortexed and transferred into dialysis tubing (Spectrum Labs, 12 kDa MMWCO). The resulting dialysis chamber was immediately placed into a vessel with 50 ml of water (recipient fluid) and incubated at 37 °C under constant and vigorous stirring. Sink conditions were maintained in the experiments, in that the concentration of MTZ in the recipient fluid at the end of the experiment did not exceed 10% of the initial drug concentration in the donor.18 During the dialysis, 0.5 ml of the recipient fluid was sampled at the following time points: 0, 1, 3, 5, 10, 20, 30, 45, 60, 90 and 120 min; and analysed for MTZ concentration. Each time the sampled volume was replaced with 0.5 ml of water. To ensure valid results, a control dialysis was performed in parallel. This was prepared by spiking the pure HAP@β-TCP suspension with the appropriate amount of MTZ prior to the standard dialysis procedure (as described above).
Ultracentrifugation method. 10 mg of powder was suspended in 50 ml of water and incubated for 120 min at 37 °C under constant magnetic stirring. During the incubation, 0.3 ml of the suspension was sampled at the following time points: 0, 1, 3, 5, 10, 20, 30, 45, 60, 90, 100, 110 and 120 min. The liquid was centrifuged (32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900× g, 5 min) and the supernatant analysed for MTZ concentration. After 90 min, 200 μl of acetic acid was added to dissolve the particles and release any possible drug residues (change of pH from 6.8 to 3.4).
900× g, 5 min) and the supernatant analysed for MTZ concentration. After 90 min, 200 μl of acetic acid was added to dissolve the particles and release any possible drug residues (change of pH from 6.8 to 3.4).
HPLC determination of metronidazole. The metronidazole concentration was determined using a Waters Alliance® HPLC system with a Waters 2695 autosampler and a Waters® 2996 Photodiode Array (PDA) detector set at 320 nm (Waters). A 150 × 2.1 mm i.d. reversed-phase column (PLRP-S 100A 5 μm, Agilent Technologies) with an appropriate guard column was used. The mobile phase comprised 90% 0.05 M CH3COONH4 at pH 4.3 and 10% acetonitrile (J. T. Baker). The flow rate was set at 0.1 mL min−1. The limit of detection (LOD) was calculated as three times the ratio of the standard deviation of the peak area in the time of elution to the slope (LOD = 3 × SD/slope), giving a value of 2.6 ng ml−1. The limit of quantification (LOQ), calculated as 10 times the ratio of the standard deviation to the slope (LOQ = 10 × SD/slope) gave a value of 8.8 ng ml−1.
2.4. Cytotoxicity assessment of pure and metronidazole-loaded HAP@β-TCP nanoparticles in macrophage (J774.E) and osteosarcoma (D17) cell lines
A cytotoxicity assessment was carried out on murine macrophage (J774.E) and canine osteosarcoma (D17) cell lines. The choice of the J774.E cell line was based on the fact that under in vivo conditions, macrophages form the primary line of response to particulate matter. Thus they are responsible for the distribution and clearance of nanoparticles and their agglomerates. The second model (D17 cells) is a cancer cell line derived from bone tissue, which is rich in hydroxyapatites that play a fundamental role in the extracellular matrix formation. Cells were cultured in a RPMI-1640 medium (Institute of Immunology and Experimental Therapy, Wrocław, Poland) supplemented with 10% fetal bovine serum (FBS, Sigma), L-glutamine (Sigma) and antibiotics (penicillin and streptomycin, Sigma). For the cytotoxicity assessment, cells were seeded in 96-well plates (NUNC) at a density of 3 × 103 (D17) or 7 × 103 (J774.E) cells per well and pre-incubated at 37 °C for 24 h in a humidified atmosphere of 5% CO2. After that, nanoparticle dispersions were added. Stock dispersions of pure HAP@β-TCP and HAP@β-TCP_MTZ were prepared based on a simplified version of the NANOGENOTOX dispersion protocol. The nanoparticles were suspended in a 0.05% BSA water solution and bath-sonicated at room temperature for 1 min. Next, the stock dispersions were further diluted in 0.05% BSA and dispersed in complete culture medium. Cells were exposed to the HAP@β-TCP and HAP@β-TCP_MTZ dispersions for 48 h (5% CO2, 37 °C). After that, the MTT assay was carried out. The test is based on the enzymatic reduction of the tetrazolium salt MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide] in living, metabolically active cells. The metabolite, purple-colored formazan, was measured colorimetrically using a multiwell plate reader. Preliminary experiments showed no interference of HAP@β-TCP or HAP@β-TCP_MTZ with either MTT or formazan in a cell-free system. After 2 h of incubation at 37 °C, 80 μl of lysis buffer was added. The buffer consisted of 225 ml dimethylformamide (Sigma); 67.5 g sodium dodecyl sulphate (Sigma) and 275 ml distilled water. The optical density (OD) was measured after 24 hours using a spectrophotometric microplate reader (ELx800, BioTek) at a wavelength of 570 nm (reference 630 nm). The OD of the control cells was taken as 100%. Cell viability was determined as follows: % viability = (mean OD in the test wells/mean OD for control wells) × 100. The results were obtained from at least 3 independent experiments.
2.5. Preparation of Ca10(PO4)6(OH)2@β-Ca3(PO4)2:Er3+/Yb3+ lobes and pellets
HAP@β-TCP lobes containing optically active ions were prepared directly from the freshly sintered and ground nanocomposites using a standard laboratory hydraulic press, applying a maximum 15 ton force on 0.1 g of powder for 60 seconds. As a result 1 mm thick lobes were obtained with diameters not exceeding 10 mm.
Spherical pellets containing HAP@β-TCP nanocomposites were prepared by vigorous mixing of 96 wt% microcrystalline cellulose (Sigma Aldrich) and 4 wt% HAP@β-TCP. After 60 s, 77.5 ml of a 20% water solution of polyvinylpyrrolidone (Sigma Aldrich) was added and the mixing process was continued, being stopped three times for the removal of residue from the walls and bottom of the container. The total mixing time was 5 min. Afterwards the mass was transferred into an extruder operating at a speed of 16 rpm and squeezed for 15 min. Subsequently, the extrudate was put into a spheronizer for 10 min at 800 rpm under a flow of compressed air. The resulting pellets were sieved and a two step drying process was applied, first at 35 °C for 5 h, and second in ambient conditions for 48 h.
2.6. Ca10(PO4)6(OH)2@β-Ca3(PO4)2:Er3+/Yb3+ nanocomposite tooth root filling
A healthy molar tooth without cracks and signs of caries needing a root filling procedure was donated by REGMED Clinic, Poland from a patient under orthodontic treatment. The tooth was thoroughly cleaned of tissue debris and blood. In order to remove the dental pulp cavity, trepanation was performed using a diamond dental bur mounted on a handpiece. Subsequently, the cavity was rinsed with 4 ml of 5.25% NaClO (Chema-Elektromet, Poland) and distilled water. The tooth roots were adjusted to 18 mm in length and the working distance was set at 0.5 mm short of the tooth apex. The root canals were explored with a 15 mm K-file (Dentsply Maillefer, Switzerland), enlarged with a 25 mm K-file (Dentsply Maillefer, Switzerland) and irrigated with 10 ml of 2% NaClO and 10 ml 0.9% NaCl after using each instrument. Afterwards, by using a Lentulo spiral, the roots and cavity were carefully filled with HAP@β-TCP paste and stored in an incubator at 37 °C with 95% humidity for 3 days. Finally, the tooth was cut using a microtome saw into two symmetrical samples. All procedures were carried out in accordance with appropriate guidelines and regulations of the Republic of Poland. The molar tooth used in the present study was obtained with the consent of the owner and according to all ethical guidelines and requirements applicable in such cases. The experiment in this study was approved by the Bioethics Committee of the Wroclaw Medical University (no. KB-525/2011).
2.7. Ca10(PO4)6(OH)2@β-Ca3(PO4)2:Er3+/Yb3+ nanocomposite deposition on implant
A basal osseointegrated type implant (BOI, IhdeDental, Germany) was covered with a HAP@β-TCP layer using a dip coating technique and further post-annealing process. Beforehand the BOI implant was washed of possible impurities and fat using an ultrasonic bath in an acetone–ethanol mixture for 25 min, and dried at 40 °C for 1 h. In order to prepare a dense water suspension of the nanocomposite, 10 g of HAP@β-TCP was taken and added into distilled water. Afterwards, the BOI was coated by a dip-coating technique and carefully transferred to a ceramic crucible for annealing at 450 °C for 3 h.
3. Results and discussion
3.1. Structural analysis of nanocomposite powders
It is well known that β-TCP crystallizes in the rhombohedral space group R3c (see Fig. 1a) having five different Ca2+ sites with coordination numbers 7 for Ca(1), 6 or 8 for Ca(2), 8 for Ca(3), 4 for Ca(4) (with partial cationic deficiency) and 6 for Ca(5), open for substitution with lanthanides.19 On the other hand, the synthetic HAP adopts a hexagonal structure depicted by the P63 space group (see Fig. 1b) with two Ca2+ sites, ninefold coordinated Ca(1) (C3) and sevenfold coordinated Ca(2) (Cs) as well.20 Since ionic radii of the Er3+ and Yb3+ cations are quite similar it can be predicted that these cations will enter most of the Ca2+ sites in both compounds.21 Therefore, complex up-conversion emission spectra are anticipated.
 |
| | Fig. 1 The projections of the crystal structures of (a) hexagonal Ca10(PO4)6(OH)2 hydroxyapatite with space group P63 (no. 173) and (b) trigonal Ca3(PO4)2 with space group R3c (no. 161). | |
The fabrication process of the HAP@β-TCP nanocomposite doped with Er3+ and Yb3+ was divided into two crucial steps. The former involved reaction pH testing, since the formation of separate β-TCP and HAP phases is driven by this parameter. Once the correct pH was found, the main aim of the latter step was to study the optimal post-treatment temperature to find a balance between sufficient crystallinity, particle size and phase content. As can be seen in Fig. 2, the synthesis carried out at acidic pH leads directly to the formation of only the β-TCP phase, whereas neutral reaction conditions result in a mixture of HAP and β-TCP. At basic pH only the HAP phase could be detected. Therefore, the effect of annealing temperature on crystal structure and phase content was studied for samples prepared at neutral pH.
 |
| | Fig. 2 The pH effect on the formation of β-TCP, HAP@β-TCP and HAP. | |
The structure evolution of the HAP@β-TCP nanocomposite doped with Er3+ and Yb3+ cations was followed by an XRD technique as a function of annealing temperature, in the range of 800–1000 °C (Fig. 3). This specific temperature regime was dictated by several factors. Firstly, it was necessary to start sintering from 800 °C since below that temperature a significant amount of residual carbon content under specific treatment conditions would be present. One could overcome this problem by extending the time of the annealing process. However, a drawback was seen in the unwanted growth of particles. Secondly, it is well known that above 1000 °C β-TCP transforms into the high temperature α-TCP phase. This means that the fabrication of nano α-TCP is questionable at such a temperature.22 Additionally, the rate of resorption of α-TCP is too quick for biological applications,23 thus the presence of α-TCP would be treated as an unwanted impurity as well.
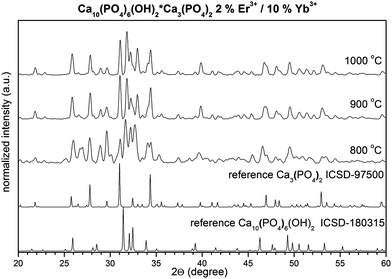 |
| | Fig. 3 Effect of annealing temperature on the structure evolution of the HAP@β-TCP nanocomposite. | |
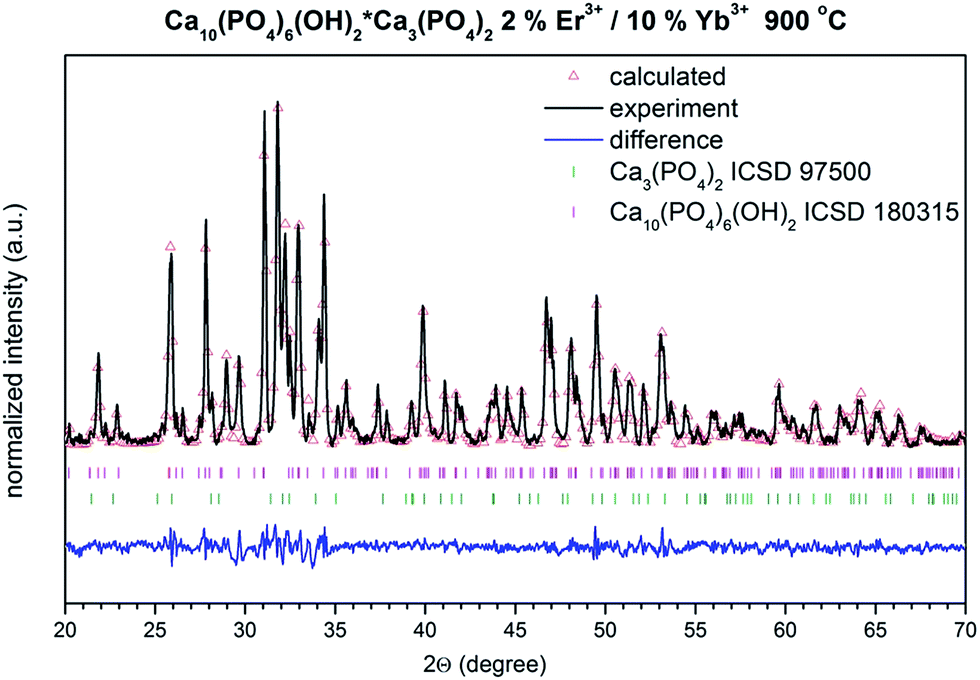
Thorough structural analysis (Fig. 4) was carried out based on the Rietveld method,24 incorporating an anisotropic approach25,26 in Maud 2.33 software.27 Fitting results are gathered in Table 1. In both cases the structural properties of HAP and β-TCP show no clear dependence on the annealing temperature. However, it is interesting to note that for the nanocomposite sintered at 800 °C, the unit cell parameters were significantly different. For instance the cell volume of the HAP phase was relatively large. It contracts above 900 °C and remained almost unchanged at 1000 °C. The reverse behavior was seen in case of the second β-TCP phase, where the cell volume expands above 900 °C, having the smallest value at 800 °C. It was quite complicated to fully comprehend this effect, but unit cell contraction could definitely be connected with the substitution of the large Ca2+ cations with smaller Er3+ and Yb3+ cations for both phases (compare with reference data). Additionally, expansion of the cell volume in the case of the HAP phase might be seen in the so-called size effect, where the action of negative pressure on the crystal lattice for small particles could lead to such results.28 Thus, it could also mean that the size of the HAP nanoparticles is smaller than those in the β-TCP phase, or that this effect is more strongly reflected by the HAP. It is worth noting that the phase content changes from 45% HAP and 54% β-TCP at 800 °C, to almost 60% HAP and 40% β-TCP above this temperature. It is well known that sintering of HAP at elevated temperature can lead to the dehydration, dehydroxylation and finally decomposition of the HAP. This process could be reversible upon the material’s contact with humidity, unless more stable phases are formed (β and/or α-TCP). Therefore, one might expect an increase in the β-TCP phase content with temperature. However, since the XRD measurement of samples heated above 800 °C was done after cooling down, the interplay of both phases is similar since water absorption might occur, leading to the hydration of β-TCP and formation of the HAP.
 |
| | Fig. 4 Representative result of the Rietveld analysis of the HAP@β-TCP doped with 2% Er3+/10% Yb3+ heated at 900 °C (black line – XRD pattern; red – fitted diffraction; blue – differential pattern; pink and green – positions of reference phase peaks). | |
Table 1 Unit cell parameters (a and c), crystal cell volume (V), as well as refined factor (Rw) for the HAP@β-TCP nanocomposite doped with 2% Er3+/10% Yb3+ as a function of sintering temperature
| Sample |
Cell parameters |
Phase |
| HAP |
β-TCP |
HAP (%) |
β-CP (%) |
Rwp (%) |
| a (Å) |
c (Å) |
V (Å3) |
a (Å) |
c (Å) |
V (Å3) |
| s. c. – single crystal reference data, HAP – ICSD 180315, β-TCP – ICSD 97500. |
| s. c.a |
9.5500 |
6.8700 |
542.62 |
10.4352(2) |
37.4029(5) |
3527.26 |
— |
— |
— |
| 800 °C |
9.5290(6) |
6.8445(1) |
538.23(6) |
10.4264(8) |
37.3453(0) |
3515.94(4) |
45 |
54 |
3.20 |
| 900 °C |
9.4255(7) |
6.8853(0) |
529.74(7) |
10.4354(2) |
37.3885(5) |
3526.05(5) |
58 |
42 |
2.52 |
| 1000 °C |
9.4287(7) |
6.8812(1) |
529.79(2) |
10.4367(1) |
37.3914(9) |
3527.20(4) |
59 |
41 |
3.50 |
Evaluation of the primary particle size and shape of the HAP@β-TCP was done by means of TEM microscopy (Fig. 5), whereas a DLS technique was employed in order to extract the hydrodynamic size of objects present in a water suspension (Fig. 6). Both techniques are of great importance in the characterization of nanoobjects, but in most cases can lead to different results. The main issue here was as follows: TEM gives only information regarding the primary size of particles and is performed on dry powders. Even though the final material showed the presence of fairly large agglomerates of particles, it was quite easy to recognize single particles and estimate their size and distribution. However, in most cases authors are not troubled by the presence of agglomerates and give only the average particle size. This is actually critical for biological applications. Thus, in order to study the behavior of particles in water or biological media it is more important to answer the question of how the state of the particles is affected by the different types of solvents and additives used for the preparation of suspensions or colloids. Therefore, the DLS technique steps into the light as the method providing more adequate or realistic results regarding the size of all objects present in the colloid.
 |
| | Fig. 5 TEM and SAED images of selected HAP@β-TCP composites prepared at 800 °C (a) and 1000 °C (b) doped with 2 mol% Er3+ and 10 mol% Yb3+. | |
 |
| | Fig. 6 Hydrodynamic size of selected Ca10(PO4)6(OH)2·Ca3(PO4)2 composite particles prepared at 800 °C doped with 2 mol% Er3+ and 10 mol% Yb3+, modified by ATP – adenosine 5′-tetrahydrogen triphosphate. | |
In accordance with the TEM analysis, the HAP@β-TCP nanocomposite sample annealed at 800 °C contains loosely agglomerated irregular particles with a primary size of 22 nm, which were starting to grow rapidly above 200 nm at 1000 °C. Since the aim of the study was to obtain a nanocomposite, further characterization was performed on samples heat treated at 800 °C.
In order to prepare a stable colloidal solution of the HAP@β-TCP nanocomposite, adenosine 5′-tetrahydrogen triphosphate (ATP) was added as a stabilizing agent, preventing agglomeration from progressing in the system over time.29 The hydrodynamic size was measured 24 and 48 h after modification of HAP@β-TCP with ATP, showing comparable values of 220 nm (Fig. 6). As one can see, this result correlates well with the TEM analysis, but only after taking into account the size of the agglomerates visible in Fig. 5a.
Actually, this is nice proof of the simple fact that once dry nanoparticles not blocked by surface agents were transferred into a water based suspension, the size estimated by these two techniques could be completely different.30 Finally, the grain size of the HAP@β-TCP nanocomposite sintered at 800 °C was verified after measuring the surface area of this sample (24 m2 g−1), giving a value of 79.36 nm. For the calculation, the sample density was taken from the Rietveld refinement, being 3.14 g cm−3. Both the TEM and BET analyses confirmed the nano-sized character of the HAP@β-TCP sample heated at 800 °C.
3.2. Metronidazole release and cytotoxicity of nanocomposites
Many inorganic nanomaterials can serve as carriers of drugs or other biologically important substances showing prospects in biomedicine applications.31 Therefore, the HAP@β-TCP nanocomposite doped with 2 mol% Er3+ and 10 mol% Yb3+ was loaded with metronidazole (an antibiotic used against anaerobic bacteria) according to the given protocol. The drug loading of HAP@β-TCP_MTZ was found to be 9.89 μg mg−1 of powder. The results of the MTZ release from the HAP@β-TCP by means of dynamic dialysis are summarized in Fig. 7.
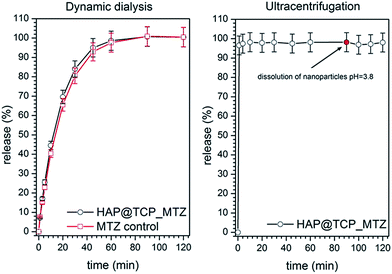 |
| | Fig. 7 Release of the metronidazole from HAP@β-TCP nanocomposite by dynamic dialysis (left) and ultracentrifugation methods (right). | |
A rapid increase in MTZ concentration in the recipient fluid has been observed during the initial 60 min of the dialysis. After this period, a plateau was reached indicating the equilibrium between the drug concentration in the donor and the recipient. The kinetics of this increase was almost identical for the drug-loaded nanoparticles and the blank HAP@β-TCP suspension spiked with MTZ. This indicates that the increase in the drug’s concentration was entirely governed by its permeation through the cellulose membrane and possible secondary interactions with suspended particles. Moreover, another conclusion might be drawn regarding weak or even lack of binding between the particles and the drug, which is even better visualised in Fig. 7. In the ultracentrifugation method, there is no barrier (i.e. dialysis membrane) that would delay the free drug molecules from entering the liquid surrounding the particles. In the present study, nearly 100% of the drug was present in the liquid instantly after the HAP@β-TCP_MTZ particles were dispersed. This confirms the lack of drug binding observed earlier with the dynamic dialysis method. Dissolution of particles after 90 min of incubation did not change the MTZ level in the solution. This excludes the presence of MTZ residues in the particle agglomerates. Thus, the HAP@β-TCP_MTZ system might be used for instance as a bifunctional material working at the same time as an immediate drug-releasing carrier in hydrophilic biological media, stimulating the treatment of bacterial infections in dental/jaw surgery and regenerative material in bone surgery.
The effects of HAP@β-TCP and HAP@β-TCP_MTZ on cell viability are summarized in Fig. 8. No significant effect of either HAP@β-TCP or HAP@β-TCP_MTZ on J774.E cell viability was observed, even at the highest concentration of 100 μg ml−1. In the case of D17 cells, the response was more variable. Metronidazole-loaded nanoparticles showed a relatively slight dose-dependent decrease in cell viability, whereas pure HAP@β-TCP did not induce any significant effect. The antiproliferative effects of pure hydroxyapatite nanoparticles towards different cancer cell lines have been described by several authors.32,33 In the present study, pure HAP@β-TCP did not show cytotoxicity, but it cannot be excluded that loading the particles with the chemotherapeutic may have triggered the cytotoxic effect due to an unknown mechanism. On the other hand, the decrease in cell viability did not reach 50% even at the highest nanoparticle concentration. Considering this, it may be concluded that neither HAP@β-TCP nor HAP@β-TCP_MTZ show significant cytotoxicity in the studied cell lines.
 |
| | Fig. 8 Mean (±SD) viability of D17 canine osteosarcoma cells (left) and J774.E murine macrophages (right) exposed for 48 h to different concentrations of pure HAP@β-TCP and nanoparticles loaded with metronidazole (HAP@β-TCP_MTZ). Viability is expressed as a percentage of the control value (results obtained from 3 independent experiments). | |
3.3. Optical properties
The absorption reflectance spectrum of the HAP@β-TCP sample containing 2 mol% Er3+ and 10 mol% Yb3+ ions heated at 800 °C was measured at 300 K in order to reveal some characteristic spectral features of both co-dopants (see Fig. 9). The spectra consist of typical absorption bands covering the UV and NIR regions associated with the intra-configurational f–f electron transition of both ions. Thus, the group of lines with sharp maxima at 379 nm (26385 cm−1) was attributed to the 4I15/2 → 4G11/2 electronic transition, that at 488.9 nm (20454 cm−1) to the 4I15/2 → 4F7/2 transition, that at 521.9 nm (19160 cm−1) to the 4I15/2 → 2H11/2 transition, and that at 1433.02 (6978 cm−1) to the 4I15/2 → 4I13/2 transition, all of these belonging to the Er3+ ions. The characteristic band with a maximum at 981 nm (10193 cm−1) was ascribed to the 2F7/2 → 2F5/2 absorption transition of the Yb3+ ions, which additionally could hinder the 4I15/2 → 4I11/2 absorption transition of the Er3+. It has to be stressed that the intensities of all the recorded bands are comparable. It is expected that this behaviour will have striking consequences for the identification of the exact up-conversion mechanism, energy transfer up-conversion (ETU) or excited state absorption (ESA). It is well known that the absorption cross section of Yb3+ (the 4F7/2 level) at 975 nm is much higher than that of Er3+ (the 4I11/2 level). Therefore, the use of additional strongly-absorbing resonant co-dopants such as Yb3+ will significantly promote the efficiency of energy transfer to the activator (Er3+).34
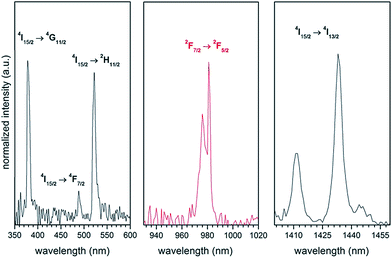 |
| | Fig. 9 Absorption reflectance spectra of the Ca10(PO4)6(OH)2·Ca3(PO4)2 composite doped with 2 mol% Er3+ and 10 mol% Yb3+, prepared at 800 °C. | |
The up-conversion emission spectra of the HAP@β-TCP sample containing 2 mol% Er3+ and 10 mol% Yb3+ ions thermally treated at 800 °C, measured at 300 K, were recorded after direct excitation at 975 nm under different laser power regimes (Fig. 10). Typical anti-Stokes emission transitions can be identified as a group of lines in the green spectral range of 505–575 nm (19801–17391 cm−1), ascribed to the 2H11/2 and 4S3/2 → 4I15/2 electronic transitions, and covering the red region of 635–689 nm (15748–14513 cm−1), attributed to the 4F9/2 → 4I15/2 electronic transition. One can note that the emission spectra are relatively broad and peaks are not resolved into specific Stark components. The most likely reason for the broadening of the emission lines is the existence of different crystallographic sites, occupied by Er3+ and Yb3+ ions in both the HAP and β-TCP structures, since all of the Er3+ ions are excited at the same time, resulting in spectral overlap of the emission lines. Another contribution could be from the presence of structural defects induced by doping with cations with different oxidation states (charge compensation effect), and/or their heterogeneous distribution due to the increased surface to volume ratio in nanomaterials. Moreover, it is worth mentioning that the intensity interplay between the two green transitions 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 changed drastically upon an increase of the laser power, in favour of the former one. The reason for this is quite simple and well-written in the literature, but has great practical implications, especially in the field of temperature sensing.35 Actually, Er3+ is one of the best candidates for the realization of such a phenomenon, since this particular ion has an appropriate energy level structure (see Fig. 11). It well known that if the considered energy levels are relatively close to each other, as in the case of 2H11/2 and 4S3/2 in Er3+, their population, represented as the integrated fluorescence intensity ratio (FIR), is driven by Boltzmann’s distribution in the following manner:
| |
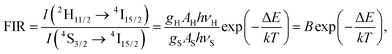 | (5) |
where
gH and
gS are the degeneracies of the
2H
11/2 and
4S
3/2 levels, respectively,
AH and
AS, and
νH and
νS are the spontaneous emission rates and frequencies of the
2H
11/2 →
4I
15/2 and the
4S
3/2 →
4I
15/2 transitions, respectively,
h is Planck’s constant,
k is Boltzmann’s constant,
T is the absolute temperature and Δ
E is the energy gap.
36 After transformation of
eqn (5) into a linear dependence:
| |
 | (6) |
the constants
B and
C can be extracted. The energy gap Δ
E equals 810 cm
−1, meaning that
C is 1165 cm
−1 (
k = 0.6952 K
−1 cm
−1). Assuming that the sample is at 300 K, the FIR can be obtained by extrapolation of its power dependence, giving a value of 0.57 at the zero limit of laser power (see inset in
Fig. 10).
37 Therefore, taking into account
eqn (6), ln(
B) is 3.32. Thus, for the highest optical laser power of 1.65 W (52.52 W cm
−2) and FIR = 1.79,
T is 425 K.
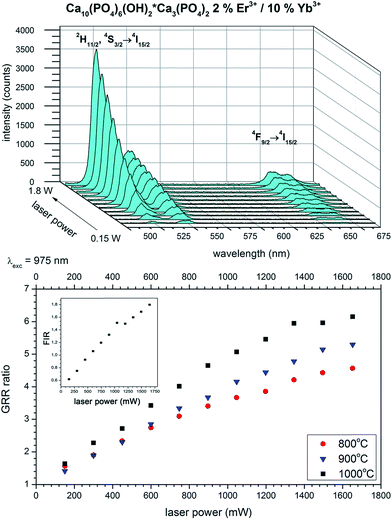 |
| | Fig. 10 Up-conversion emission spectra as a function of laser power (top) and GRR (bottom), together with FIR power dependence (inset) of the Ca10(PO4)6(OH)2·Ca3(PO4)2 composite doped with 2 mol% Er3+ and 10 mol% Yb3+, prepared at 800 °C. | |
It is interesting to note that the intensity of the green part of the spectra strongly increased over the red region with an increase of laser power as well. Analysis of the integrated intensity ratio between the green transitions and the red ones could be treated as an indirect method for estimation of the efficiency of the up-conversion process (green-to-red ratio GRR).38 The general rule is as follows, if the red band intensity increases at the expense of the green bands, the efficiency of the up-conversion process decreases due to the enhanced role of the non-radiative processes (cross-relaxation, multiphonon relaxation). Therefore, the higher the GRR value, the better the performance of the up-converting system. This simple method could be the basis for comparison of different materials. The GRR is expected to be dependent on the type of phase in terms of symmetry of occupied sites, since the closest surrounding influence is splitting in the crystal field, as well as being dependent on excitation power, inducing thermal effects and directly influencing the population of electronic states.39 One can clearly see (Fig. 10 right) that the GRR of the HAP@β-TCP nanocomposite doped with 2 mol% Er3+ and 10 mol% Yb3+ strongly increased upon an increase in laser power from a low power regime of 5.7 W cm−2 (0.18 W) where the GRR is 1.4, to a higher power regime, where the GRR is 6.2 at 57.3 W cm−2 (1.8 W). This change was definitely caused by the sample self-heating and the thermalization process, resulting in higher population of 2H11/2 → 4I15/2. It has to be emphasised that the GRR also depends on the grain size of particles. The highest values were achieved for the largest particles, since the smaller ones contain a higher fraction of optically active cations located closer to the surface, which are prone to non-radiative deactivation (impurities, surface states, defects etc.).
In the case of the up-conversion emission, three main mechanisms were proposed for the APTE (addition de photon par transfer d’energie) effect, named also as energy transfer up-conversion (ETU), excited state absorption (ESA) and photon avalanche (PA).40 In the majority of cases ETU and ESA were the most effective. However, due to the quadratic power dependence it was difficult to differentiate between them. A schematic energy-level diagram showing the mechanism of up-conversion and the accompanying processes is shown in Fig. 11. After absorption of NIR photons by both Yb3+ and Er3+, electrons from the ground states 2F7/2 and 4I15/2 were excited to the 2F5/2 and 4I11/2 levels by the ground state absorption (GSA) process instantaneously. Additional incoming photons moved electrons from the 4I11/2 level to the 4F7/2 level by excited state absorption (ESA).
 |
| | Fig. 11 Simplified energy level scheme showing the up-conversion and cross relaxation (CR) processes. | |
Due to the large absorption cross section of Yb3+ ions, the majority of the excitation energy was absorbed by Yb3+, and electrons at the 2F5/2 level could be directly transferred to the 4I11/2 level of the Er3+ by ET (energy transfer) and/or to the 4F7/2 level of Er3+ by ETU. Both ETU and ESA demand the participation of two photons. Afterwards, electrons from the 4F7/2 level relaxed very fast non-radiatively to the 2H11/2 and 4S3/2 levels via the multiphonon relaxation (MPR) process. From these levels, radiative de-excitation could occur to the 4I15/2 level, resulting in green emission eventually. Since there is always competition between radiative and non-radiative depopulation, some of the electrons could be lost on feeding of the 4F9/2 level due to the multiphonon relaxation process of the 2H11/2 + 4S3/2 levels, or cross relaxation (CR). This behaviour could be tuned by careful selection of host lattice, balance of co-dopant concentration, grain size and synthetic parameters. The CR is strongly concentration- and laser power-sensitive. One can identify the following several highly probable CR processes:
| | |
(2H11/2 + 4S3/2, 4I15/2) → (4I9/2, 4I13/2)
| (I) |
| | |
(2H11/2 + 4S3/2, 4I13/2) → (4F9/2, 4I11/2)
| (II) |
| | |
(4S3/2, 4I9/2) → (4F9/2, 4F9/2)
| (III) |
| | |
(4S3/2, 4I13/2) → (4I9/2, 4I9/2)
| (IV) |
| | |
(4I11/2, 4I11/2) → (4I15/2, 4F7/2)
| (V) |
| | |
(4I11/2, 4F7/2) → (4F9/2, 4F9/2)
| (VI) |
all of them contributing to a decrease in the green emission and GRR. Additionally, since the energy gaps between the
4S
3/2 and
4F
9/2 and the
4F
9/2 and
4I
9/2 levels are equal to 3000 and 2850 cm
−1, respectively, the emission may be effectively quenched by phonons from vibrations of the
ν4 PO
43− and
ν3 PO
43− phosphate groups, or surface defects like OH
− bridging the energy mismatch. This behavior may also suggest that ESA dominates over ETU, which would be also confirmed by less efficient up-conversion intensity in comparison to other materials, such as fluorides for example.
34
The pump power dependence of the green (2H11/2, 4S3/2 → 4I15/2) and red (4F9/2 → 4I15/2) emissions was investigated (see Fig. 12) as a function of sintering temperature, for samples containing 2 mol% Er3+ and 10 mol% Yb3+. The experimental results were fitted with a linear function, giving slope values of the green band close to 2 and around 1 for the red band, confirming the involvement of two photons in green emission. The situation was somewhat different in the case of the red emission. Actually, the 4F9/2 level could be fed by at least three main processes (1) nonradiative decay from the 2H11/2 and 4S3/2 levels, (2) through ETU involving the 4I13/2 level and (3) cross relaxations II, III, and VI. As stated by Pollnau,41 if the nonradiative decay processes dominate, both the slopes of green and red emission tend to be close to 2. In the case when the 4F9/2 level is populated from 4I15/2 through ETU, the slope of power dependence of the red band should be close to 3 (three photon process).39 In the studied case, the value of the power dependence of the 4F9/2 → 4I15/2 transition was close to 1, pointing to the increasing role of cross relaxation processes, as similarly stated by Liu.42
 |
| | Fig. 12 Power dependence of the Ca10(PO4)6(OH)2·Ca3(PO4)2 composite doped with 2 mol% Er3+ and 10 mol% Yb3+, prepared at 800 °C. | |
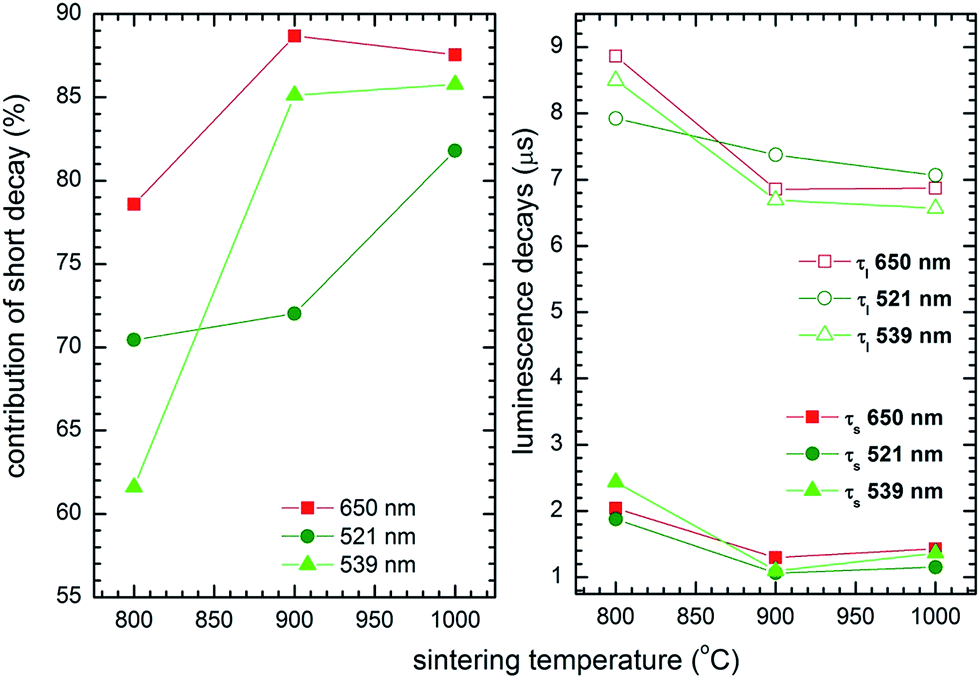
The luminescence decay was multiexponential and exhibited short and long decay components (Fig. 13) equal to 1.44 ± 0.59 μs and 6.5 ± 2.3 μs at 654 nm, 1.48 ± 0.4 μs and 7.45 ± 0.3 μs at 521 nm, and finally 1.8 ± 0.1 μs and 7.5 ± 1.0 μs at 539 nm. The double exponent behavior may be rationalized by two possible mechanisms. The first one could originate from surface effects; Er3+ ions located within the aggregate core exhibit longer luminescence lifetimes, while those located at the surface are more susceptible to the local environment. As a consequence these show shorter luminescence lifetimes. However, significant structural changes have been found as the annealing conditions were changed. While the sample annealed at 800 °C showed a significant contribution from the β-TCP phase and differed from the samples annealed at higher temperatures, with the elevation of annealing temperature the short decay component contribution seemed to increase, as well as an overall decrease in both the short and long components of the decay time (see Fig. 14). It has to be mentioned that in classical examples of up-converting materials, the radiative decay times of both green and red transitions in mixed metal oxides are usually of the order of a few hundred microseconds in low concentration samples. Both components being fast, especially upon comparison with bulk materials, and a lack of visible rise times could imply the presence of enhanced nonradiative transfers due to the presence of Er3+ surface-rich regions and multiple Er3+ ions close to each other, due to the nano-size character of the particles in the former case, and the relatively high concentration of Er3+ in the latter. It is well known that a size reduction below the micrometer scale results in detrimental effects, reducing up-conversion intensity and efficiency.43,44
 |
| | Fig. 13 Luminescence decay curves of the Ca10(PO4)6(OH)2·Ca3(PO4)2 nanocomposite doped with 2 mol% Er3+ and 10 mol% Yb3+, prepared at 800 °C. | |
 |
| | Fig. 14 Short decay component contribution (left) as well as values of short and long decay components (right) as a function of sintering temperature of the Ca10(PO4)6(OH)2·Ca3(PO4)2 nanocomposite doped with 2 mol% Er3+ and 10 mol% Yb3+. | |
In the case of relatively highly concentrated compounds, it is expected that donor–donor energy transfer would be efficient, leading to the concentration quenching effect.45 In general energy is transferred to the traps and dissipated in the crystalline net due to the overlap of f orbital wave functions of RE3+ through the oxygen lattice band. Since the Er3+ and Yb3+ ions are replacing Ca2+ ions in the Ca10(PO4)6(OH)2·Ca3(PO4)2 composite, it was logically expected that this would induce the formation of vacancies and enhance such nonradiative processes. Furthermore, one can clearly see that the decay time of the red emission is a few times longer than that of the green emission. This might be a straightforward indication of strong nonradiative CRs depopulating the green bands (as indicated in Fig. 11).
3.4. Potential application of up-converting HAP@β-TCP nanocomposite in bio-medicine
The main aim of this section is to show the application prospects of the developed system containing the HAP@β-TCP nanocomposite optically activated with up-converting Er3+ and Yb3+ co-dopants. As can be easily found in the literature, biphasic HAP and TCP have shown very good osteoconductive potential,46 which is the main issue in bone tissue regeneration. However, we would like to propose different forms and uses of the HAP@β-TCP Er3+/Yb3+ nanocomposite: lobes, pellets, tooth root filling material, or a composite for covering titanium-based implants (see Fig. 15 and 16), directly taking advantage of this type of material in bio-related applications.
 |
| | Fig. 15 Different forms of the HAP@β-TCP Er3+/Yb3+ up-converting nanocomposite: lobes and pellets (left), tooth root filler (right); before and after excitation with NIR irradiation. | |
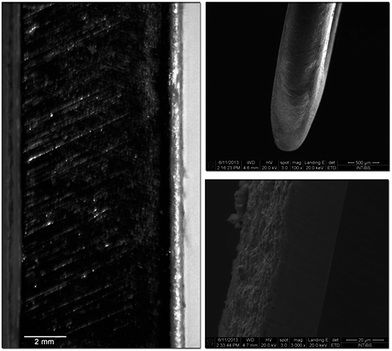 |
| | Fig. 16 Cross section of a titanium BOI implant covered with a layer of the HAP@β-TCP Er3+/Yb3+ up-converting nanocomposite (left), side head view of the implant (upper right) and the layer/implant boundary (bottom right). | |
Application of the HAP@β-TCP Er3+/Yb3+ lobes, characterized by different size and thickness and covered with collagen membranes, could promote bone healing processes especially in the field of dental surgery and implantology.
Pellets tailored to a specific size with optimal porosity serving as carriers of antibiotics such as metronidazole could be used as bone substitute materials in orthopedic and craniofacial surgery due to their high osteoconductive properties. The up-converting lanthanide cations within the nanocomposite might be utilized as bio-markers and specific indicators of bone redevelopment as well as the fate of the biomaterial in terms of its re-mineralization, re-absorption and the presence of individual particles in the site of application. The root filling HAP@β-TCP Er3+/Yb3+ nanocomposite additionally loaded with antibiotics and steroids due to its biocompatibility could be used as a temporary or end filling material, especially when the lower pH at the inflammation site promotes the release of Ca2+ ions from nanomaterials, which are responsible for endothelium sealing and the generation of the phosphatase enzyme. Furthermore, the HAP@β-TCP activated with Er3+/Yb3+ ions could be used for tooth labelling and studies of the remineralization or rebuilding of the dentin and/or enamel. Finally, BOI-type titanium implants could be easily covered with a desired layer thickness to improve their biocompatibility and biointegration within the bone tissue. As can be seen, the structure of the outer layer is porous and this could be used for carrying anti-inflammatory substances to minimize the risk of after-surgery complications or to speed up the healing process. The possibility of bio-imaging of the HAP@β-TCP Er3+/Yb3+ outer layer might be useful in the evaluation of layer/implant ageing, and to follow the integration of the implant with bone tissue by observation of the ion diffusion upon possible re-build of the boundary tissue. Colloids containing up-converting particles might be used in IR-thermometry or for direct destruction of unwanted cells, utilizing thermal effects by raising the particle temperature above 40 °C when it is already located at its specific destination.
4. Conclusions
Up-converting HAP@β-TCP nanocomposites activated with Er3+/Yb3+ ion pairs were successfully fabricated using Pechini’s technique, with the phase ratio being close to 60:40 depending on the annealing conditions. The primary size of the particles was evaluated by TEM and was found to be 22 nm in the case of the lowest temperature treatment, whereas the hydrodynamic size was around 220 nm and did not change significantly after time upon ATP stabilization. It has been shown that HAP@β-TCP modified with metronidazole could be used as a carrier for immediate drug release, since there is only very weak bonding of the metronidazole to the surface of the proposed system. The cytotoxicity of the nanocomposite was tested on canine osteosarcoma (D17) cells, showing a slight concentration-dependent effect on cell viability, whereas no effect was observed in the case of murine macrophage J774.E cells. It was found that the interplay of the intensity between the 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions changes upon sample heating induced by different laser powers, giving a strong base for the use of this material in emission thermometry. Analysis of the GRR led to the conclusion that an increase in the GRR with laser power is due to the thermalization process, resulting in higher population of 2H11/2 → 4I15/2. It was found that the GRR depends strongly on the grain size of the particles. Large particles contain a lower fraction of optically active cations located close to surface areas and are thus less prone to non-radiative deactivation. An energy level diagram was shown for a detailed description of the up-conversion and accompanying processes.
The recorded luminescence decay was multiexponential and exhibited short and long decay components with rather short decay values in all cases. The double exponential behavior was rationalized by the presence of surface effects, meaning that Er3+ ions located within the aggregate core exhibit longer luminescence lifetimes, while those located at the surface are more susceptible to the local environment. Additionally, relatively fast components of decay time as well as a lack of rise times imply an enhanced contribution of nonradiative transfers. This is mostly due to the presence of Er3+ surface-rich regions, due to the nano-size character of the particles. However, the existence of multiple Er3+ ions close to each other because of the relatively high concentration of Er3+ cannot be excluded as well. Therefore, it is expected that donor–donor energy transfer would be efficient, leading to the concentration quenching effect.
Acknowledgements
The authors would like to thank Ewa Bukowska M.Sc. for performing XRD measurements, as well as Malgorzata Malecka Ph.D. for TEM images and Dorota Kida M.Sc. for pellet preparation. REGMED Clinic, Poland is gratefully acknowledged for the donation of teeth. Financial support from the National Science Centre over the course of the realization of the project ‘Preparation and characterization of nanoapatites doped with rare earth ions and their biocomposites’, no. UMO-2012/05/E/ST5/03904, is gratefully acknowledged.
References
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413–3431 CrossRef CAS PubMed.
- H. Liu and T. J. Webster, Biomaterials, 2007, 28, 354–369 CrossRef CAS PubMed.
- L. L. Hench and J. Wilson, Science, 1984, 226, 630–636 CAS.
- Z. Y. Li, W. M. Lam, C. Yang, B. Xu, G. X. Ni, S. A. Abbah, K. M. C. Cheung, K. D. K. Luk and W. W. Lu, Biomaterials, 2007, 28, 1452–1460 CrossRef CAS PubMed.
- O. Kozo, T. Yamamuro, T. Nakamura and T. Kokubo, Biomaterials, 1990, 11, 265–271 CrossRef.
- D. K. Chatteriee, A. J. Rufalhah and Y. Zhang, Biomaterials, 2008, 29, 937–943 CrossRef PubMed.
- S. A. Hilderbrand, F. Shao, C. Salthouse, U. Mahmood and R. Weissleder, Chem. Commun., 2009,(28), 4188–4190 RSC.
- M. P. Ferraz, A. Y. Mateus, J. C. Sousa and F. J. Monteiro, J. Biomed. Mater. Res., Part A, 2007, 81A, 994–1004 CrossRef CAS PubMed.
- E. Landi, A. Tampieri, G. Celotti, S. Sprio, M. Sandri and G. Logroscino, Acta Biomater., 2007, 3, 961–969 CrossRef CAS PubMed.
- I. Salma, G. Salms, M. Pilmane, D. Loca, J. Locs, N. Romanchikova, A. Skagers and L. Berzina Cimdina, Int. J. Oral Maxillofacial Surg., 2011, 40, 1216 CrossRef PubMed.
- M. E. Fleet and Y. M. Pan, J. Solid State Chem., 1994, 112, 78–87 CrossRef CAS.
- L. D. DeLoach, S. A. Payne, L. L. Chase, L. K. Smith, W. L. Kway and W. F. Krupke, IEEE J. Quantum Electron., 1993, 29, 1179–1191 CrossRef CAS.
- K. Spariosu, R. D. Stultz, M. Birnbaum, T. H. Allik and J. A. Hutchinson, Appl. Phys. Lett., 1993, 62, 2763 CrossRef CAS PubMed.
- R. J. Wiglusz, A. Bednarkiewicz and A. Lukowiak, Spectrosc. Lett., 2010, 43, 333–342 CrossRef CAS.
- R. J. Wiglusz, A. Kedziora, A. Lukowiak, W. Z. Doroszkiewicz and W. Strek, J. Biomed. Nanotechnol., 2012, 8, 605–612 CrossRef CAS PubMed.
- J. Rao, A. Dragulescu-Andrasi and H. Yao, Curr. Opin. Biotechnol., 2007, 18, 17–25 CrossRef CAS PubMed.
- Z. Li, Y. Zhang and S. Jiang, Adv. Mater., 2008, 20, 4765–4769 CrossRef CAS.
- Y. Zambito, E. Pedreschi and G. Di Colo, Int. J. Pharm., 2012, 434, 28–34 CrossRef CAS PubMed.
- M. Yashima, A. Sakai, T. Kamiyama and A. Hoshikawa, J. Solid State Chem., 2003, 175, 272–277 CrossRef CAS.
- E. Balan, S. Delattre, D. Roche, L. Segalen, G. Morin, M. Guillaumet, M. Blanchard, M. Lazzeri, C. Brouder and E. K. H. Salje, Phys. Chem. Miner., 2011, 38, 111–122 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- Y. C. Fung, Biomechanics. Mechanical Properties of Living Tissues, Springer-Verlag Inc., New York, 1993, p. 500 Search PubMed.
- J. B. Park, Biomaterials Science and Engineering, Plenum Press, New York, 1987 Search PubMed.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- R. Delhez, T. H. de Keijser, J. I. Langford, D. Louër, E. J. Mittemeijer and E. J. Sonneveld, Crystal Imperfection Broadening and Peak Shape in the Rietveld Method, in The Rietveld Method, ed. R. A. Young, Oxford Science, Oxford, United Kingdom, 1993, p. 132 Search PubMed.
- L. Luterotti and P. Scardi, J. Appl. Crystallogr., 1990, 23, 246–252 CrossRef.
- L. Lutterotti, S. Matthies and H.-R. Wenk, IUCr: Newsl. CPD, 1999, 21, 14–15 Search PubMed.
- V. R. Palkar, P. Ayyub, S. Chattopadhyay and M. Multani, Phys. Rev. B: Condens. Matter, 1996, 53, 2167–2170 CrossRef CAS.
- R. Pązik, R. Anderson, L. Kępiński, V. G. Kessler and G. A. Seisenbaeva, J. Phys. Chem. C, 2011, 115, 9850–9860 Search PubMed.
- R. C. Murdock, L. Braydich-Stolle, A. M. Schrand, J. J. Schlager and S. M. Hussain, Toxicol. Sci., 2008, 101, 239–253 CrossRef CAS PubMed.
- R. K. Singh and H. W. Kim, J. Tissue Eng. Regener. Med., 2013, 10, 296–309 CrossRef CAS.
- W. Tang, Y. Yuan, C. Liu, Y. Wu, X. Lu and J. Qian, Nanomedicine, 2014, 9, 397–412 CrossRef CAS PubMed.
- Y. Yuan, C. Liu, J. Qian, J. Wang and Y. Zhang, Biomaterials, 2010, 31, 730–740 CrossRef CAS PubMed.
- H. Schafer and M. Hasse, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef PubMed.
- M. L. Debasu, D. Ananias, I. Pastoriza-Santos, L. M. Liz-Marzan, J. Rocha and L. D. Carlos, Adv. Mater., 2013, 25, 4868–4874 CrossRef CAS PubMed.
- T. V. Gavrilovic, D. J. Jovanovic, V. Lojpur and M. D. Dramicanin, Sci. Rep., 2014, 4, 4209 Search PubMed.
- B. Dong, S. Xu, J. Sun, S. Bi, D. Li, X. Bai, Y. Wang and L. Wang, J. Mater. Chem., 2011, 21, 6193 RSC.
- K. W. Kramer, D. Biner, G. Frei, H. U. Gudel, M. P. Hehlen and S. R. Luthi, Chem. Mater., 2004, 16, 1244–1251 CrossRef.
- J. F. Suyver, J. Grimm, K. W. Kramer and H. U. Gudel, J. Lumin., 2005, 114, 53–59 CrossRef CAS PubMed.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- M. Pollnau, D. R. Gamelin, S. R. Luthi and H. U. Gudel, Phys. Rev. B: Condens. Matter, 2000, 61, 3337–3346 CrossRef CAS.
- L. Liu, H. Jiang, Y. Chen, X. Zhang, Z. Zhang and Y. Wang, J. Lumin., 2013, 143, 423–431 CrossRef CAS PubMed.
- F. Auzel and D. C. Pecile, C. R. Seances Acad. Sci., Ser. B, 1973, 277, 155 CAS.
- J. Silver, M. I. Martinez-Rubio, T. G. Ireland, G. R. Fern and R. J. Withnall, J. Phys. Chem. B, 2001, 105, 948 CrossRef CAS.
- L. G. Van Uitert, R. C. Linares, R. R. Soden and A. A. Ballman, J. Chem. Phys., 1962, 36, 702 CrossRef PubMed.
- B. D. Boyan and Z. Schwartz, Nat. Rev. Rheumatol., 2011, 7, 8–9 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.