
Enhanced fast charge–discharge performance of Li4Ti5O12 as anode materials for lithium-ion batteries by Ce and CeO2 modification using a facile method
Abstract
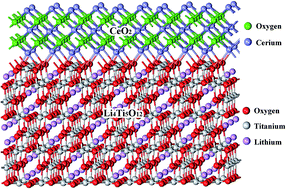
A facile solid-state method to improve the fast charge–discharge and kinetic performance of Li4Ti5O12 in lithium-ion batteries by Ce and CeO2 in situ modification is presented in this work. XRD shows that the Ce doping and CeO2 modification do not change the spinel structure of Li4Ti5O12. Little Ce doping (Ti/Ce = 4.9 : 0.1 and Ti/Ce = 4.85 : 0.15) reduces the lattice parameter of doped Li4Ti5O12, but more Ce4+ doping (Ti/Ce = 4.8 : 0.2) increases the lattice parameter due to the large ionic radius of Ce4+. Raman spectra reveal that CeO2 is not completely incorporated into the host structure and leads to the formation of a uniform coating on the surface of Li4Ti5O12. The doping of Ce4+ and the combination with in situ generated CeO2 in Li4Ti5O12 are favorable for reducing the electrode polarization and charge-transfer resistance and improve the lithium insertion/extraction kinetics of Li4Ti5O12, resulting in its relatively higher capacity at a high charge–discharge rate. The Ce-doped Li4Ti5O12–CeO2 composites show a much improved rate capability and cycling stability compared with pristine Li4Ti5O12 at a 10 C charge–discharge rate in a broad voltage window (0–2.5 V). The introduction of Ce and CeO2 enhances not only the electric conductivity of Li4Ti5O12, but also the lithium ion diffusivity in Li4Ti5O12, resulting in a significantly improved high-rate capability, cycling stability, and fast charge–discharge performance of Li4Ti5O12.
 Please wait while we load your content...
Please wait while we load your content...

