DOI:
10.1039/C5RA00441A
(Paper)
RSC Adv., 2015,
5, 20081-20089
Efficient catalytic activation of Suzuki–Miyaura C–C coupling reactions with recyclable palladium nanoparticles tailored with sterically demanding di-n-alkyl sulfides†
Received
9th January 2015
, Accepted 2nd February 2015
First published on 3rd February 2015
Abstract
n-Bromodocosane reacts with Na2S, generated in situ by the reduction of elemental sulfur with NaBH4, to give n-didocosyl sulfide (L1), which acts as a protector for palladium nanoparticles (2–7) that are prepared using different palladium precursors in the presence of L1 (Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L1 ratio 1:2 and 4:1). The NPs have been characterized with powder X-ray diffraction, SEM, EDX, UV-vis spectroscopy and HRTEM. The size (nm) ranges for the majority of spherical NPs 2–7 are ∼18–19, 4–5, 5–7, 4–6, 7–9 and 4–6 respectively. The precursor of palladium affects the size, shape and dispersion of the NPs. When [Pd(CH3CN)2Cl2]/Na2PdCl4 was used as a precursor, uniformly dispersed NPs of narrow size range were obtained. L1 and its complex [Pd(L1)2Cl2] (1) have also been synthesized by the reaction of Na2PdCl4 with L1 and characterized with 1H and 13C{1H} NMR spectroscopy. The NPs show good catalytic activity for the Suzuki–Miyaura coupling (SMC) of various aryl chlorides/bromides with phenylboronic acid at low catalyst loading (0.1–0.5 mol% of Pd). The conversion is good for some aryl halides in a short reaction time of the order 1–2 h. Among 2–7, the highest activity is observed for Pd NPs obtained from Na2PdCl4, which is probably due to uniformity in their size and dispersion. The distinct advantage of NPs 2–7 is that they can be separated and reused at least up to five times. The complex 1, equivalent to 0.001 mol% Pd, is efficient for the SMC of some aryl halides, as good conversion into coupled products has been observed. Two phase tests, conducted for 1 and 3, suggest the contribution of both homogeneous and heterogeneous catalytic pathways in overall catalysis.
:L1 ratio 1:2 and 4:1). The NPs have been characterized with powder X-ray diffraction, SEM, EDX, UV-vis spectroscopy and HRTEM. The size (nm) ranges for the majority of spherical NPs 2–7 are ∼18–19, 4–5, 5–7, 4–6, 7–9 and 4–6 respectively. The precursor of palladium affects the size, shape and dispersion of the NPs. When [Pd(CH3CN)2Cl2]/Na2PdCl4 was used as a precursor, uniformly dispersed NPs of narrow size range were obtained. L1 and its complex [Pd(L1)2Cl2] (1) have also been synthesized by the reaction of Na2PdCl4 with L1 and characterized with 1H and 13C{1H} NMR spectroscopy. The NPs show good catalytic activity for the Suzuki–Miyaura coupling (SMC) of various aryl chlorides/bromides with phenylboronic acid at low catalyst loading (0.1–0.5 mol% of Pd). The conversion is good for some aryl halides in a short reaction time of the order 1–2 h. Among 2–7, the highest activity is observed for Pd NPs obtained from Na2PdCl4, which is probably due to uniformity in their size and dispersion. The distinct advantage of NPs 2–7 is that they can be separated and reused at least up to five times. The complex 1, equivalent to 0.001 mol% Pd, is efficient for the SMC of some aryl halides, as good conversion into coupled products has been observed. Two phase tests, conducted for 1 and 3, suggest the contribution of both homogeneous and heterogeneous catalytic pathways in overall catalysis.
Introduction
Palladium catalysts are widely used for the Suzuki–Miyaura coupling (SMC), which is a powerful synthetic tool in organic chemistry for C–C bond formation.1 They include both molecular and nano sized species.2,3 Though the catalysis is carried out by palladium(0) in the case of both Pd nanoparticles (NPs) and molecular complexes, the role of the ligand present appears to be crucial as it affects the efficiency of the catalyst. Therefore, phosphorus, carbene, oxime and several other ligands have been used to design molecular complexes of palladium, which are suitable as efficient catalysts for SMC.4–12 Generally, these complexes do not catalyze the coupling but act as dispensers of real palladium(0) containing catalytic species (via nano-sized palladium13 if formed in the course of catalysis). When palladium NPs are used as catalysts for this coupling reaction, their surface area and morphology are important considerations for catalytic performance. The high surface-to-volume ratio and increase in atomic distribution on the surface are expected to enhance the catalytic activity. The synthesis of well-defined and mono-dispersed (small size range) Pd NPs continues to be a challenge. The ligands used to protect Pd NPs may reduce the catalytic activity. Thus, the protection strategy for NPs should be such that the activity loss is minimal.14 Note that the role of the protecting ligand is very crucial because it determines the dispersion of the NPs and affects their catalytic efficiency, recyclability, composition, solubility and size.15 Various agents, such as dendrimers, organic ligands, organofluorous compounds, ionic liquids and surfactants, have been found to protect metal NPs.2,4,16 The presence of a functional group, such as thiol, amine, carbonyl or hydroxyl, in a protector limits the activity15 of NPs due to strong chemisorptions. Recently, some organochalcogen ligands, mainly alkyl thiolates, have been utilized as protectors for designing catalytically active transition metal (such as Pd, Pt and Au) NPs.16–18 The thiolate protected Pd and Pt NPs that have been reported as catalysts for organic reactions include palladium NPs protected with alkylthiolate for the catalysis of Suzuki–Miyaura coupling14b,17e and isomerization of allyl alcohol,17f–h as well as Pt NPs, which are protected with alkylthiol and alkyl amine and have good potential for methanol oxidation and hydrogenation of allyl alcohol and maleic acid, respectively.17a–c,h Hydrogenation of allylamine has been successfully catalyzed by cyclodextrin thiolate protected Pt and Pd nanoparticles.17d Surprisingly, thioethers, which are soft donors, less toxic and easy to handle in air, have been slightly explored as protectors to design catalytically active metal NPs. Recently, Obare and coworkers have reported the hydrogenation of styrene using Pd NPs protected with a thioether ligand.18 However, predictable control of particle size with ligands continues to be a challenge.18 The precursor of metal and protecting ligand together may be envisaged as a suitable combination for controlling, to some extent, the properties of NPs, including catalytic activity. The bulky thioether ligand used as a protector may result in good dispersion and, in turn, an active nano-sized metal catalyst. Herein, we report the Pd NPs prepared using different Pd precursors and didocosyl sulfide [CH3(CH2)20CH2–S–CH2–(CH2)20CH3] (L1) as a protecting ligand. Their catalytic performance for SMC has been studied and found to be promising; moreover, they are found to be recyclable. The synthesis of the palladium(II) complex [PdCl2(L1)2] and its activity to catalyze SMC are also reported; moreover, the complex is also an efficient catalyst (couples some ArBr at a Pd loading level of as low as 0.0001 mol%). It activates coupling via formation of Pd based NPs. Note that the Pd precursors affect size, shape, and dispersion of Pd NPs, which in turn affect their catalytic activity.10,15b All these results are described in this paper.
Experimental section
Physical measurement
1H and 13C{1H} spectra were recorded on a Bruker Spectrospin DPX 300 NMR spectrometer at 300.13 and 75.47 MHz, respectively. The chemical shifts are reported in ppm relative to the internal standard tetramethylsilane. 13C DEPT NMR was used to routinely determine the number of hydrogen atoms linked to various carbon atoms. Elemental analyses were carried out with a Perkin-Elmer 2400 Series II C, H, N analyzer. HRTEM studies were carried out with a Tecnai G2 20 electron microscope operated at 200 kV. The specimens for this purpose were prepared by dispersing the powdered solid in ethanol by ultrasonication, dropping slurry onto a porous carbon film supported on a copper grid and then drying it in air. Carl Zeiss EVO5O scanning electron microscope (SEM) equipped with an EDX system model QuanTax 200, based on the SDD technology was used to observe elemental composition of the nanoparticles. The samples for SEM were mounted on a circular metallic sample holder with a sticky carbon tape and scanned in different regions in order to minimize the error in the analysis made for evaluating the morphological parameters. Powder X-ray diffraction (PXRD) studies were carried out on a Bruker D8 advance diffractometer with Ni-filtered CuKα radiation using a scan speed of 1 s and scan step of 0.02°. Shimadzu UV2450 spectrophotometer with UV probe software has been used to record UV-vis spectra. Melting points of the ligand and complex were determined in an open capillary sealed at one end with an apparatus equipped with electric heating and reported as such. The products of catalytic reactions were separated and purified (if required) by chromatography using silica gel (60–120 mesh) column. n-Hexane and its mixtures with chloroform/ethyl acetate in variable proportions were used as eluents. The products were authenticated by matching their NMR data with those reported in the literature. Commercially available reagents were used as received. All the reactions were carried out in an oven-dried round bottom flask under ambient conditions. Before use, commercial nitrogen gas was successively passed through traps containing solutions of alkaline anthraquinone–sodium dithionite, alkaline pyrogallol, and concentrated H2SO4 and KOH pellets. Nitrogen atmosphere, if required, was created using Schlenk techniques. An atomic absorption spectrophotometer (AAS), Lab India AAS 7000, was used to determine Pd content in NPs. 10, 20, and 30 ppm solutions of Pd(II) prepared from Na2PdCl4 in deionised water were used as standards.
Starting materials
Sulfur powder, NaBH4, 1-bromodocosane, sodium tetrachloropalladate, palladium acetate, palladium(II) chloride, potassium carbonate, caesium carbonate, phenylboronic acid and all aryl halides were procured from Aldrich (USA). Bis(acetonitrile)dichloropalladium(II) was prepared by refluxing palladium chloride in acetonitrile.
Syntheses of L1 (n-C22H45)2S. Sulfur powder (0.06 g, 2.0 mmol) was stirred in dry ethanol (30 mL) for 1 h at 70 °C. Aqueous saturated solution of NaBH4 containing NaOH (0.20 g) was added dropwise to it under a nitrogen atmosphere. After the reaction mixture turned greenish yellow, solid 1-bromodocosane (1.55 g, 4 mmol) was added with stirring. Thereafter, the reaction mixture was stirred further at 60 °C for 8 h. On completion of the reaction, the precipitate was filtered, washed with ethanol (3 × 30 mL) and dried in vacuo. Yield: white solid; (1.11 g) 85%; m.p. 68 °C. Anal. found: C, 79.74%; H, 13.26%. Calc. for C44H90S: C, 79.85%; H, 13.33%. 1H NMR (300 MHz, CDCl3, 25 °C, TMS); δ (ppm): 0.88 (t, J = 6.6 Hz, 6H, H22), 1.25–1.56 (m, 80H, H2, H3–H21), 2.49 (t, J = 76.9 Hz, 4H, H1). 13C{1H} NMR (75 MHz, CDCl3, 25 °C, TMS); δ (ppm): 14.1 (C22), 22.7 (C21), 28.5–29.7 (C20–C4), 31.9 (C3), 32.1 (C2), 39.3 (C1).
Synthesis of complex 1. Na2[PdCl4] (0.147 g, 0.5 mmol) was dissolved in 10 mL of methanol. The solution of ligand L1 (0.65 g, 1 mmol) in a mixture of chloroform and hexane (20 mL each) was added to it with vigorous stirring. The mixture was further stirred for 2 h. Thereafter, the volume of the reaction mixture was reduced to 10 mL on a rotary evaporator. The resulting yellow precipitate was filtered, washed with methanol (3 × 20 mL) and dried in vacuo. Yield: light yellow solid (0.59 g) 80%; m.p. 78 °C(d). Anal. found: C, 70.27%; H, 11.81%. Calc. for Pd[Cl2S(C22H45)2]: C, 70.42%; H, 11.76%. 1H NMR (300 MHz, CDCl3, 25 °C, TMS); δ (ppm): 0.88 (m, 12H, H22), 1.25–1.86 (m, 152H, H21–H3), 1.40–1.86 (m, 8H, H2), 2.67–2.78 (m, 8H, H1). 13C{1H} NMR (75 MHz, CDCl3, 25 °C, TMS); δ (ppm):14.1 (C22), 22.6 (C21), 28.2 (20), 28.9–29.7 (C19–C4), 30.9 (C3), 31.9 (C2), 36.1 (C1).
Synthesis of palladium NPs 2–7. Pd(OAc)2 (0.224 g, 1.0 mmol) for 2 and 5/Na2[PdCl4] (0.294 g, 1.0 mmol) for 3 and 6/Pd[(CH3CN)2Cl2] (0.259 g, 1.0 mmol) for 4 and 7 was dissolved in 50 mL of methanol. The solution of ligand L1 [1.30 g, 2 mmol (Pd:L1 = 1:2) for 2, 3 and 4/0.162 g, 0.25 mmol for 5, 6 and 7 (Pd:L1 = 4:1)] prepared in 100 mL of a mixture of chloroform and hexane (equal volumes of each) was added to the abovementioned solutions with vigorous stirring. The reaction mixture was further stirred for 12 h at room temperature. For NPs 3, 4, 6 and 7, the solution of NaBH4 (0.111 g, 3.0 mmol) in methanol was added additionally dropwise to the reaction mixture with stirring over a period of 30 min. Methanol acts as a reducing agent for palladium acetate in the synthesis of NPs 2/5. On the completion of reaction, the solvent in the mixture was reduced to 10 mL on a rotary evaporator. The resulting residue was centrifuged, washed with methanol (3 × 30 mL) and dried in vacuo.
Procedure for Suzuki–Miyaura C–C coupling reaction catalyzed with 1
An oven-dried round bottom flask was charged with aryl halide (1 mmol), phenylboronic acid (1.2 mmol), K2CO3 (2 mmol) and DMF/H2O (3 mL/1 mL). An appropriate amount of complex 1 as a solution made in chloroform was added. The flask was placed on an oil bath at 100 °C. Progress of the reaction was monitored by 1H NMR. After the completion of the reaction, the product was extracted with 10 mL of diethyl ether. The organic phase was washed with water (3 × 10 mL) and dried over anhydrous Na2SO4. The solvent was removed on a rotary evaporator and the residue was subjected to 1H and 13C{1H} NMR. Further purification, if required, was performed by column chromatography on silica gel using hexane/ethyl acetate mixture (90:10) as an eluent.
Procedure for the Suzuki–Miyaura C–C coupling reaction catalyzed with nanoparticles (2–7)
An oven-dried round bottom flask was charged with aryl halide (5 mmol), phenylboronic acid (6 mmol), K2CO3 (8 mmol) and DMF/H2O (9 mL/3 mL). Nanoparticles (one from 2–7) were added (amount equivalent to 0.2 mol% of Pd in case of 2–4 and 0.5 mol% in case of 5–7). The reaction mixture was heated on an oil bath, and the temperature was maintained at 100 °C. The progress of the reaction was monitored with NMR. On the completion of the reaction, the product was extracted with 10 mL of diethylether. The extract was washed with water (3 × 10 mL) and dried over anhydrous Na2SO4. The solvent of the extract was evaporated off and the residue was subjected to NMR.
Catalytic recyclability for NPs
An oven-dried round bottom flask was charged with aryl bromide (5 mmol), phenylboronic acid (6 mmol), K2CO3 (8 mmol) and DMF/H2O (9 mL/3 mL). Nanoparticles (one from 2–4) were added (amount equivalent to 0.2 mol% of Pd). The reaction mixture was heated on an oil bath, and the temperature was maintained at 100 °C. The progress of reaction was monitored with NMR. After maximum conversion, the reactants were added in the same proportion to the original reaction mixture to monitor the catalytic recycling. Conversions were estimated with 1H NMR (Table 6). After the completion of the fifth catalytic cycle, the reaction mixture was cooled to room temperature. The coupled product was extracted with 20 mL diethylether or ethyl acetate (in case of 4). The extract was washed with water (3 × 20 mL) and dried over anhydrous Na2SO4. The solvent in the extract was evaporated off and residue was subjected to NMR to authenticate the product.
Catalytic recyclability of NPs after separation
The coupling reaction of 4-bromobenzoic acid was carried out with NPs 4 (0.2 mol% of Pd). After the completion of the reaction, ethyl acetate and water were added to the reaction mixture and it was centrifuged. Aqueous-organic layer was decanted and the black residue that was obtained was washed thoroughly with ethyl acetate and water to remove organic content and base, respectively. The resulting residue was dried in vacuo. The organic layer from ethyl acetate–water system was separated. Its solvent was evaporated and residue was subjected to 1H NMR for estimating the conversion. The NPs separated were reused for catalysis using an optimized procedure. This procedure was repeated five times and results are given in Table 8.
Isolation of Pd NPs generated from 1 in coupling reaction
An oven-dried round bottom flask was charged with 4-bromobenzaldehyde (1 mmol), phenylboronic acid (1.2 mmol), K2CO3 (2 mmol), DMF/H2O (3 mL/1 mL) and large amount of complex 1 (200 mg). The flask was placed on an oil bath and the temperature was maintained at 100 °C. On the completion of the reaction, the black particles settled down in the reaction mixture. The aqueous-organic layer was decanted, and the black residue that was obtained was centrifuged and thoroughly washed with ethyl acetate and water to remove the base and organic content. Then, it was washed again with acetone–water (20 + 10 mL). The resulting residue was dried in vacuo, and HRTEM study revealed that the residue was in the form of spherical NPs (Fig. 1g).
 |
| | Fig. 1 HRTEM images of nanoparticles. (a) 2 (scale bar 100 nm), (b) 3 (scale bar 20 nm), (c) 4 (scale bar 20), (d) 5 (scale bar 50 nm), (e) 6 (scale bar 50 nm), (f) 7 (scale bar 20 nm), (g) NPs obtained in SMC of complex 1 (scale bar 20 nm), (h) NPs 6 after a catalytic reaction (scale bar 50). | |
Hg poisoning test
Excess of Hg (Hg:Pd; 400:1) was taken in a reaction flask before the addition of reactants. Thereafter, the coupling reaction of 4-bromobenzaldehyde (1 mmol) with phenylboronic acid (1.2 mmol) using 1/3 (0.1/0.2 mol% of Pd) as a catalyst under optimal conditions was carried out. After carrying out the reaction for 12 h, the cross-coupled derivative for 1 and 3 was obtained in 100% and 67% yield, respectively.
PPh3 poisoning test
To the coupling reaction of 4-bromobenzaldehyde (1 mmol) with phenylboronic acid (1.2 mmol), PPh3 (5.2 mg, 2 mol%) was added under optimal conditions. Thereafter, 1/3 (equivalent to 0.1/0.2 mol% of Pd) was added to the reaction mixture. After 18 h of reaction, the cross-coupled product 4-formylbiphenyl was obtained in ∼100% yield in both the cases.
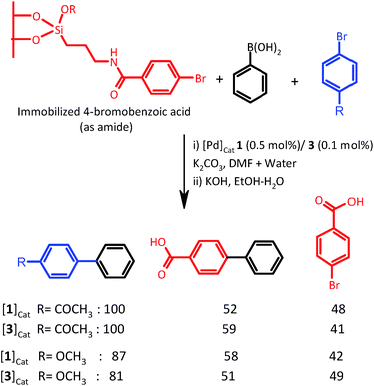
Two/three-phase test
A mixture of 4-bromobenzoic acid-immobilized silica (0.20 g), prepared using a reported procedure,19 phenylboronic acid (0.36 g, 3 mmol), 4-bromoacetophenone or 4-bromoanisole (1 mmol) and K2CO3 (0.56 g, 4 mmol) was heated at 100 °C for 12 h in a mixture of DMF/water (6 mL/3 mL) in the presence of 1/3 (0.5/0.1 mol%). Thereafter, the reaction mixture was cooled and filtered through a G-4 crucible. The residue was washed with 30 mL of water followed by diethyl ether (40 mL). The filtrate and washings were collected together, and the resulting mixture was extracted with 30 mL of diethyl ether. The solvent of the extract was evaporated on a rotary evaporator and the residue was subjected to 1H NMR. The residue in G-4 crucible was hydrolysed with KOH (1.68 g dissolved in 10 mL of EtOH + 5 mL of H2O) at 90 °C for 3 days. The hydrolysed solution was neutralized with 20% (v/v) aqueous HCl and extracted with dichloromethane (30 mL) followed by ethyl acetate (40 mL). The organic phases were combined together and the solvent was evaporated. Then, the resulting residue was analyzed with 1H NMR.
Results and discussion
Synthesis and characterization
The syntheses of L1, its Pd(II)–complex 1 and Pd NPs 2–7 (protected with L1) are summarized in Scheme 1. Note that these compounds are stable in air; moreover, the high protection of NPs, which is provided by L1, is revealed by the fact that centrifugation of their dispersions in chloroform at 6000 rpm does not result in any aggregation (see ESI; Fig. S29†). The air insensitive L1 and diamagnetic palladium complex 1 have been characterized using their 1H and 13C{1H} NMR spectra. These spectra (see ESI; Fig. S1–S4†) are consistent with their structures depicted in Scheme 1. The Pd NPs 2–7 have been synthesized (Scheme 1) using different palladium precursors in varying metal to ligand ratios. The Pd:L1 ratio is 1:2 for 2, 3 and 4 and 4:1 for 5, 6 and 7; moreover, the palladium precursors Pd(OAc)2, Na2PdCl4 and [Pd(CH3CN)2Cl2] have been used in case of 2/5, 3/6 and 4/7, respectively (Scheme 1). All these NPs (2–7) have been subjected to powder XRD (see ESI Fig. S20–S25†), HRTEM (Fig. 1), SEM and SEM-EDX (see ESI Fig. S5–S10 and S12–S17†) for characterization. The peaks at d ∼ 2.28 (hkl: 111) and 1.97 (hkl: 200) observed in the powder XRD of 6 and 7 (see ESI Fig. S24 and S25†) support the presence of palladium(0) (JCPDS ≠ 88-2335) with face-centered cubic structure. Because of high content of protector ligand (see SEM-EDX results), these peaks do not appear in case of 2, 3 and 4. The peaks present at 2.318° and 4.131° in powder XRD of ligand were found slightly shifted in powder XRD of 2, 3 and 4 (see ESI Fig. S20–S22†), indicating the interaction of ligand with NPs. The SEM-EDX studies have revealed that Pd:S ratios (in atomic%) in NPs 2–4 are 35:65, 39:61 and 33:67, respectively. This ratio in case of 5, 6 and 7 has been found to be 40:60, 61:39 and 74:26, respectively. The size (nm) ranges for NPs (majority) of Pd are ∼18–19, 4–5, 5–7, 4–6, 7–9 and 4–6 respectively for 2–7. The size distribution graphs given in ESI (Fig. S26–S28†) suggest that the pattern of growth varies for nanoparticles 2 to 7. In the case of 2, the reduction of Pd(OAc)2 is slow as it is caused by MeOH, a mild reducing agent. Thus, the reduction time is prolonged, causing multi-nucleation and uneven growth rate and has resulted in big Pd NPs with wider size range.15b Comparison of the size of 2 and 5 reveals that it may also be contributed by high ligand:Pd(OAc)2 ratio. Probably, the reduction is slowed down due to increase in number of ligand molecules in the reaction mixture, which in turn may lead to further aggregation during the growth process. The large size in case of NPs 2 leads to their lower catalytic activity in comparison to that of 3 and 4 (Table 2). The NPs 3, 4, 6 and 7 have been synthesized by reduction with sodium borohydride, a strong reducing agent, over a period of 30 min. Their small size and good dispersion result is due to the fast reduction process. The bulky and soft donor site containing ligands around Pd NPs are known to prevent their aggregation during growth.15b Thus, the quality of NPs (size and dispersion) is determined by a balance between rate of reduction and control of ligand over growth. In the reduction process, Pd(II) is fully converted into Pd(0) as revealed by the UV-vis spectrum. There is no peak at 235 nm due to the presence of Pd(II)/[PdCl4]2− in the dispersion of NPs. The appearance of the peak at around 305 nm due to Pd(0) along with a characteristic peak of ligand L1 at ∼250 nm (Fig. 2) in the UV-vis spectrum of each NP indicates a weak interaction between the ligand and the surface of palladium of NPs.15c
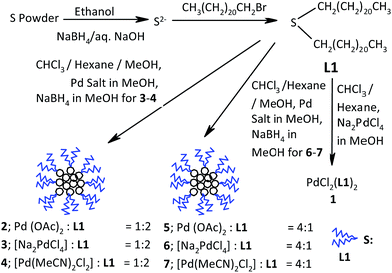 |
| | Scheme 1 Synthesis of L1, Pd(II) complex 1 and Pd nanoparticles 2–7. | |
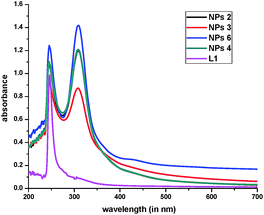 |
| | Fig. 2 UV-vis spectrum of NPs 2–4, 6 and L1. | |
Catalysis
The complex 1 and NPs 2–7 have been explored for SMC of several aryl halides, including heteroaryl bromides and aryl chlorides. To optimize the reaction conditions, the coupling reaction of 4-bromobenzaldehyde with phenylboronic acid in the presence of 1 was studied (Table 1) using various bases and solvents. K2CO3 has been found to be the most appropriate base because the yield of cross-coupled product is lower compared to alternatives such as caesium carbonate, sodium acetate, potassium hydroxide, and triethylamine. Furthermore, the reaction was influenced by solvents. A mixture of DMF and water was found to give the best results (Table 1: entry 5). Complex 1 was found to show catalytic activity for aryl chlorides (Table 2: entries 1 and 2) but at higher catalyst loading (1.0 mol% of Pd). For aryl bromides, good catalytic conversions were obtained with 1 even at its very low loading (10−4 mol% of Pd). Moreover, air insensitivity of 1 is a distinct advantage. The extent of variation in catalytic activity of 1 for aryl chlorides and bromides was revealed when the coupling reaction of 4-chlorobenzaldehyde under optimized reaction conditions (Table 2: entry 2) was compared with that of 4-bromobenzaldehyde (Table 2: entries 6 and 7). 89% yield of the cross-coupled product (Table 2: entry 2) was obtained in 40 h using 1 mol% of catalyst loading in the case of 4-chlorobenzaldehyde, whereas 4-bromobenzaldehyde gave yield of same order (Table 2: entries 7) within 9 h at a very low catalyst loading (10−4 mol% of Pd). The effect of change in catalyst loading on cross-coupled product formation was studied for 4-bromobenzaldehyde, 1-bromo-4-nitrobenzene and 4-bromoanisole. The yield of the coupled product in the case of 4-bromobenzaldehyde remained almost the same (Table 2: entries 6, 7) on varying the catalyst loading between 10−2 and 10−4 mol% if the reaction time was slightly increased for the lower loading. The yield did not vary significantly on varying the catalyst loading from 0.1 to 10−3 and from 1.0 to 0.5 mol% of Pd in the case of 1-bromo-4-nitrobenzene and 4-bromoanisole, respectively, if the reaction time was adjusted. The reaction time required for the completion of the reaction varied (Table 2: entries 4, 5, 10 and 11) significantly from 16 to 1 and 6 to 3 h on increasing the catalyst loading in the case of 1-bromo-4-nitrobenzene and 4-bromoanisole, respectively. The efficiency of 1 is also reflected in high conversion (92%; Table 2: entries 10 and 11) of 4-bromoanisole, which is an electronically deactivated substrate. In the course of coupling reaction between 4-bromobenzaldehyde and phenylboronic acid catalyzed with 1, the formation of black particles was observed. Thus, this reaction was carried out using a larger amount of 1 (see Experimental) to obtain good quantity of black particles for further investigations. These black particles were isolated, washed thoroughly, dried and characterized with HRTEM, SEM and SEM-EDX. HRTEM studies have revealed that these particles are of nanosize (∼4 nm). They are catalytically active (Table 7) due to their protection with sulfur ligands20,21 but are required in higher amount to provide sufficient Pd loading for good conversion in comparison to that of precursor molecular complex. The presence of protecting ligands with black particles is supported by SEM-EDX, which reveals that NPs contain Pd as well as sulfur, and the approximate Pd:S ratio (in atom%) is 28:72.
Table 1 Optimization of reaction conditionsa
| CHO–C6H4–Br + PhB(OH)2 → CHO–C6H4–Ar |
| Entry no. |
Solvent |
Base |
Yieldb (%) |
| Reaction conditions: 1 mmol of 4-bromobenzaldehyde, 1.2 mmol of phenylboronic acid, 2 mmol of base, 5 mL of solvent, temperature of bath 100 °C, 0.0001 mol% of 1 and reaction time was 10 h. NMR yield. |
| 1 |
DMF + Water |
Cs2CO3 |
91 |
| 2 |
DMF + Water |
CH3COONa |
67 |
| 3 |
DMF + Water |
KOH |
82 |
| 4 |
DMF + Water |
NEt3 |
10 |
| 5 |
DMF + Water |
K2CO3 |
96 |
| 6 |
DMF |
K2CO3 |
81 |
| 7 |
Toluene |
K2CO3 |
11 |
| 8 |
THF |
K2CO3 |
29 |
| 9 |
DMSO |
K2CO3 |
78 |
Table 2 Suzuki–Miyaura coupling reactions catalyzed by 1a
| Ar–X + PhB(OH)2 → Ar–Ph (Ar: aryl/heteroaryl; X: Cl/Br) |
| Entry no. |
Aryl/heteroaryl halide |
mol% |
t (h) |
Yieldb (%) |
| Reaction conditions: 1 mmol of aryl/heteroaryl halide, 1.2 mmol of phenylboronic acid, 2 mmol of K2CO3, DMF + H2O (3 + 1 mL), temperature of bath 100 °C. Isolated yield. |
| 1 |
1-Chloro-4-nitrobenzene |
1 |
23 |
94 |
| 2 |
4-Chlorobenzaldehyde |
1 |
40 |
89 |
| 3 |
4-Bromobenzonitrile |
0.1 |
4 |
93 |
| 4 |
1-Bromo-4-nitrobenzene |
0.1 |
1 |
96 |
| 5 |
1-Bromo-4-nitrobenzene |
10−3 |
16 |
94 |
| 6 |
4-Bromobenzaldehyde |
10−2 |
7 |
95 |
| 7 |
4-Bromobenzaldehyde |
10−4 |
9 |
94 |
| 8 |
4-Bromoacetophenone |
10−2 |
9 |
92 |
| 9 |
4-Bromotoluene |
0.1 |
14 |
91 |
| 10 |
4-Bromoanisole |
1 |
3 |
92 |
| 11 |
4-Bromoanisole |
0.5 |
6 |
90 |
| 12 |
2-Bromopyridine |
0.1 |
10 |
90 |
The results of investigations on catalytic activities of NPs 2–7 for SMC are given in Tables 3, 4 and 5. 2–7 have been explored for the reaction of 4-bromoanisole with phenylboronic acid under optimum reaction conditions (Table 3). The order of efficiency has been found as 3 > 5 > 4 > 6 > 2 > 7 (Table 3). The cross-coupled product was obtained in ∼100% yield after the reaction of 1 h in case of 3. These results show that metal:ligand ratio and the precursor used in the synthesis of NPs affect their catalytic activity. The NPs 2 show low catalytic efficiency, which may be due to their large size as evident by their TEM image (Fig. 1a). The TEM image recorded after the catalytic reaction of 4-bromoanisole with phenylboronic acid in the presence of 6 (Table 3) suggests that the quality of NPs 6 does not degrade and that they remain well dispersed even after the reaction (see Fig. 1e and h). The activity of NPs 2–7 has been studied further for various aryl halides. The NPs 2 and 3 show almost similar reactivity towards the coupling reaction of 4-chlorobenzaldehyde/1-chloro-4-nitrobenzene. However, NPs 4 do not show any catalytic activity towards aryl chloride substrates. The results given in Tables 4 and 5 reflect that 2–7 can be successfully used to couple several aryl bromides in 1 to 4 h with their low loading (0.2–0.5 mol% of Pd). Generally, the product obtained, using these catalysts is pure; therefore, column chromatography is not required for further purification. The recyclability of 2, 3 and 4 was studied for the coupling reaction of 4-bromobenzaldehyde, 4-bromoanisole and 4-bromobenzoic acid, respectively, with phenyl boronic acid. 2 and 4 do not show any decrease in their efficiencies for the reaction of 4-bromobenzaldehyde and 4-bromobenzoic, respectively, (Table 6) even after the fifth reaction cycle. However, the catalytic activity of 3 was reasonably decreased for the coupling reaction of 4-bromoanisole after the third reaction cycle (Table 6). To check whether these NPs are active after their isolation from the reaction mixture, coupling of 4-bromobenzoic acid with phenyl boronic acid under optimal reaction conditions was carried out by reusing the separated NPs 4. Results shown in Table 8 indicate that the separated NPs are active even after the fourth catalytic cycle. HRTEM of these NPs recorded after the first catalytic cycle (Fig. 3) does not show significant change in their size and distribution. The NPs 2–7 protected with L1 are required in an amount (in terms of mol% of Pd) higher than that of 1 for the same SMC reaction in order to get comparable conversion but they are recyclable. The good efficiency of 2–7 may be attributed to the large surface-to-volume ratio resulting from the absence of any direct covalent interaction between protecting ligand and surface palladium atoms. In earlier studies, the weak binding between metal atoms on the surface of NPs and ligands has been found to result in well dispersed nanoparticles of high catalytic reactivity.15a
Table 3 Activity of Pd NPs 2–7 for Suzuki coupling of 4-bromoanisolea
| |
Catalyst |
| 2 |
3 |
4 |
5 |
6 |
7 |
| Reaction conditions: 4-bromoanisole (1 mmol), phenyl boronic acid (1.2 mmol), K2CO3 (2 mmol), DMF + H2O (3 + 1 mL), temperature of bath 100 °C, time 1 h, amount of NPs 2–7 equivalent of 0.1 mol% of Pd. NMR yield. |
| Yieldb (%) |
71 |
∼100 |
85 |
90 |
72 |
Trace |
Table 4 Suzuki–Miyaura C–C coupling reactions catalyzed with Pd NPsa 2–4
| Ar–X + PhB(OH)2 → Ar–Ph (X: Cl/Br) |
| Entry no. |
Aryl halide |
2 |
3 |
4 |
| t (h) |
Yieldb |
t (h) |
Yieldb |
t (h) |
Yieldb |
| Reaction conditions: ArX (5 mmol), phenylboronic acid (6 mmol), K2CO3 (8 mmol), DMF + H2O (9 + 3 mL), temperature of bath 100 °C, amount of 2, 3 and 4 equivalent to 0.2 mol% of palladium. NMR % yield. Amount of catalyst equivalent to 1.0 mol% of palladium. |
| 1c |
4-Chlorobenzaldehyde |
20 |
91 |
20 |
95 |
20 |
16 |
| 2c |
1-Chloro-4-nitrobenzene |
20 |
∼100 |
20 |
∼100 |
24 |
Trace |
| 3 |
4-Bromobenzaldehyde |
2 |
97 |
2 |
∼100 |
2 |
∼100 |
| 4 |
4-Bromobenzoic acid |
4 |
94 |
4 |
94 |
4 |
∼100 |
| 5 |
1-Bromo-4-nitrobenzene |
2 |
96 |
2 |
∼100 |
2 |
∼100 |
| 6 |
4-Bromobenzonitrile |
3 |
89 |
3 |
93 |
3 |
∼100 |
| 7 |
4-Bromotoluene |
4 |
91 |
4 |
95 |
4 |
85 |
| 8 |
4-Bromoanisole |
4 |
87 |
1 |
∼100 |
4 |
96 |
| 9 |
2-Bromopyridine |
8 |
— |
11 |
86 |
8 |
95 |
Table 5 Suzuki–Miyaura C–C coupling reactions catalyzed with Pd NPsa 5–7
| Ar–Br + PhB(OH)2 → Ar–Ph |
| Entry no. |
Aryl bromide |
5 |
6 |
7 |
| t (min) |
Yieldb |
t (min) |
Yieldb |
t (min) |
Yieldb |
| Reaction conditions: ArBr (5 mmol), phenylboronic acid (6 mmol), K2CO3 (8 mmol), DMF + H2O (9 + 3 mL), temperature of bath 100 °C, amount of 5, 6 and 7 equivalent to 0.5 mol% of Pd. NMR % yield. |
| 1 |
4-Bromobenzaldehyde |
90 |
∼100 |
90 |
∼100 |
90 |
∼100 |
| 2 |
Bromobenzene |
90 |
91 |
90 |
∼100 |
90 |
∼100 |
| 3 |
4-Bromoanisole |
90 |
87 |
90 |
∼100 |
90 |
∼100 |
Table 6 Recyclability of NPs in Suzuki–Miyaura C–C coupling reactionsa
| Ar–Br + PhB(OH)2 → Ar–Ph |
| Entry no. |
Aryl bromide |
|
Reaction cycle |
| 1 |
2 |
3 |
4 |
5 |
| Reaction conditions: ArBr (5 mmol), phenylboronic acid (6 mmol), K2CO3 (8 mmol), DMF + H2O (9 + 3) mL, temperature of bath 100 °C. Amount of 2–4 equivalent to 0.2 mol% of Pd. NMR % yield. |
| 1 |
4-Bromobenzaldehydeb |
t (h) |
4 |
4 |
4 |
4 |
4 |
| Yieldc |
∼100 |
∼100 |
∼100 |
∼100 |
∼100 |
| 2 |
4-Bromoanisoleb |
t (h) |
1 |
1 |
1 |
1 |
1 |
| Yieldc |
∼100 |
∼100 |
∼100 |
85 |
81 |
| 3 |
4-Bromobenzoic acidb |
t (h) |
5 |
5 |
5 |
5 |
5 |
| Yieldc |
∼100 |
∼100 |
∼100 |
∼100 |
∼100 |
Table 7 Suzuki–Miyaura C–C coupling reactions catalyzed by Pd NPs obtained from complex 1a
| Ar–Br + PhB(OH)2 → Ar–Ph |
| Entry no. |
Aryl bromide |
NPs obtained from 1 |
| t (min) |
Yieldb (%) |
| Reaction conditions: ArBr (1 mmol), phenyl boronic acid (1.2 mmol), K2CO3 (2 mmol), DMF + H2O (3 + 1 mL), temperature of bath 100 °C. Amount of NPs equivalent to 0.5 mol% of Pd (calculated by AAS). NMR yield. |
| 1 |
4-Bromobenzaldehyde |
90 |
∼100 |
| 2 |
Bromobenzene |
90 |
87 |
| 3 |
4-Bromobenzoic acid |
90 |
91 |
Table 8 Recyclability of NPs 4 in Suzuki–Miyaura C–C coupling reactiona
| COOH–C6H4–Br + PhB(OH)2 → COOH–C6H4–Ar |
| Entry no. |
Aryl bromide |
|
Reaction cycle |
| 1 |
2 |
3 |
4 |
5 |
| Reaction conditions: 4-bromobenzoic acid (1 mmol), phenylboronic acid (1.2 mmol), K2CO3 (2 mmol), DMF + H2O (3 + 1 mL), temperature of bath 100 °C. Amount of 4 equivalent to 0.2 mol% of Pd. NMR % yield. |
| 1 |
4-Bromobenzoic acid |
t (h) |
5 |
5 |
5 |
5 |
5 |
| Yieldb |
∼100 |
∼100 |
∼100 |
95 |
92 |
 |
| | Fig. 3 HRTEM image of NPs 4 after first catalytic cycle. | |
To establish the pathway of catalysis with molecular complex 1 and NPs 2–7, mercury and triphenylphosphine poisoning and two phase tests were performed. The coupling of 4-bromobenzaldehyde was carried out in the presence of 1/3 (0.1/0.2 mol% of Pd) for the mercury poisoning test. After 12 h of reaction time, the cross-coupled product for 1 and 3 was obtained in ∼100 and 67% yield, respectively. Thus, catalysis by molecular complex 1 is not quenched by Hg. However, the catalysis by NPs has been found to be diminished to some extent. The coupling reaction of 4-bromobenzaldehyde activated with 1/3 was also smooth in the presence of triphenylphosphine because the product was formed in almost in ∼100% yield after 18 h of reaction. Thus, catalysis is not heterogeneous with 1. Because Pd NPs are protected with ligand molecules, the possibility of a heterogeneous pathway cannot be presumed. On the basis of mercury and triphenylphosphine poisoning tests alone, one cannot convincingly conclude the nature of the catalytic process. Thus, a two/three-phase test (Scheme 2) was carried out to ascertain the contributions of heterogeneous and homogeneous catalytic processes.19 This test provides a better idea about catalytically active metal species.
 |
| | Scheme 2 Two phase test in the presence of 1 and 3. | |
Note that this test was developed by Rebek and co-workers.22 In this test, a coupling partner (aryl halide) is heterogenized and the second coupling partner (phenylboronic acid) along with another aryl halide (homogeneous) have been reacted in the presence of catalyst under optimum reaction conditions. If the catalyst behaves in a heterogeneous manner, the supported aryl halide is not expected to be converted to a coupled product. When palladium is released into a solution, the supported substrate is likely to be converted into a product. Thus, as shown in Scheme 2, 4-bromobenzoic acid immobilized silica was reacted with phenylboronic acid in the presence of 4-bromoacetophenone or 4-bromoanisole under optimum reaction conditions using 1 and 3 as catalysts. After 12 h of reaction time, the soluble part was separated and analyzed with 1H NMR. The yield of the cross-coupled product was ∼100% for 4-acetylbiphenyl and 81–87% for 4-methoxybiphenyl. The solid phase separated through G-4 crucible was hydrolyzed, and the resulting residue after workup was analyzed with 1H NMR. In the presence of 4-bromoacetophenone, the conversion of immobilized 4-bromobenzoic to biphenyl-4-carboxylic acid was observed to be 52% when complex 1 was used as a catalyst. The conversion was 59% when NPs 3 were used as a catalyst. In the presence of 4-bromoanisole, the conversions of immobilized substrate for catalysts 1 and 3 were 58% and 51%, respectively. The relative rates of reaction of immobilized and free ArBr are not of much significance in concluding the nature of catalysis. The absolute conversion of immobilized substrate indicates whether catalysis is homogeneous or heterogeneous. The present results indicate that in both the cases, the reaction is homogeneously as well as heterogeneously catalyzed with Pd species. However, the contribution of homogeneous catalysis is marginally higher. It is possible that 1 behaves as a precatalyst and generates nano-sized particles of palladium, which contributes to a heterogeneous pathway. In the case of palladium NPs, it could be concluded that the catalytically active palladium leached from NPs is responsible for homogeneous catalysis. Thus, the catalysis is neither purely homogeneous nor purely heterogeneous and can be described as “cocktail-like”.21,23–25
Conclusions
Palladium NPs 2–7 have been prepared using n-didocosyl sulfide (L1) as a protector and different precursors of palladium in different ratios with respect to ligand L1. The precursor of palladium affects the size, shape and dispersion of NPs. The size (nm) ranges for NPs (majority) of Pd are: 2: ∼18–19; 3: ∼4–5; 4: ∼5–7; 5: ∼4–6; 6: ∼7–9; 7: ∼4–6. The NPs have been characterized using powder X-ray diffraction, SEM, EDX, UV-vis and HRTEM. The complex [Pd(L1)2Cl2] (1) has also been synthesized and characterized with 1H and 13C{1H} NMR spectroscopy. The NPs show good catalytic efficiency for the Suzuki–Miyaura coupling (SMC) of various aryl halides with phenylboronic acid at a low catalyst loading (0.1–0.5 mol% of Pd). The conversion is good for some aryl halides even in a short reaction time of the order 1–2 h. The size and dispersion were most favourable for catalytic activity when Na2PdCl4 was used as a precursor. The distinct advantage associated with the NPs 2–7 is that they remain active in solution even after the reaction and also after their separation; moreover, they can be reused up to five times. The complex 1 is also efficient for SMC as its loading equivalent to 0.001–0.0001 mol% Pd is optimum for good conversion of some aryl bromides into coupled products. A two phase test, conducted in the presence of 1 and 3, suggests the contribution of homogeneous as well as heterogeneous Pd species in the catalytic processes.
Acknowledgements
Council of Scientific and Industrial Research and Department of Science and Technology, India, supported the work through projects [CSIR: 01(2784)14/EMR-II; DST: SR/S1/IC-40/2010]. SK, MPS and FS thank UGC for the fellowship. GKR and AK thank CSIR for the fellowship. Authors thank Professor A. K. Ganguli for providing the HR-TEM facility at IIT Delhi.
Notes and references
-
(a) Metal-Catalyzed Cross-Coupling Reaction, ed. A. de Meijere and F. Diederich, Wiley- VCH, Weinheim, 2004 Search PubMed;
(b) Metal-Catalyzed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998 Search PubMed;
(c) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi and A. de Meijere, Wiley-Interscience, New York, 2002, p. 1669 Search PubMed.
-
(a) F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 2419 CAS;
(b) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed;
(c) H. Li, C. C. C. J. Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147 CrossRef CAS;
(d) D. Yuan and H. V. Huynh, Molecules, 2012, 17, 2491 CrossRef CAS PubMed;
(e) N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048 CrossRef CAS PubMed.
-
(a) A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181 RSC;
(b) D. Astruc, Inorg. Chem., 2007, 46, 1884 CrossRef CAS PubMed;
(c) N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef CAS;
(d) D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS PubMed;
(e) A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS PubMed.
-
(a) J. P. Wolfe and S. L. Buchwald, Angew. Chem., Int. Ed., 1999, 38, 2413 CrossRef CAS;
(b) A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020 CrossRef CAS;
(c) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS;
(d) X. Bei, H. W. Turner, W. H. Weinberg and A. S. Guram, J. Org. Chem., 1999, 64, 6797 CrossRef CAS PubMed.
- R. B. Bedford and C. S. J. Cazin, Chem. Commun., 2001, 1540 RSC.
- R. B. Bedford, S. L. Hazelwood and M. E. Limmert, Chem. Commun., 2002, 2610 RSC.
- S. Teo, Z. Weng and T. S. A. Hor, Organometallics, 2006, 25, 1199 CrossRef CAS.
- C. A. Fleckenstein and H. Plenio, Organometallics, 2007, 26, 2758 CrossRef CAS.
- J. Ma, X. Cui, L. Gao and Y. Wu, Inorg. Chem. Commun., 2007, 10, 762 CrossRef CAS PubMed.
- J. P. Wolfe, R. A. Singer, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 9550 CrossRef CAS.
-
(a) C. Fleckenstein, S. Roy, S. Leuthaeusser and H. Plenio, Chem. Commun., 2007, 2870 RSC;
(b) A. I. Moncada, J. M. Tanski and L. M. Slaughter, J. Organomet. Chem., 2005, 690, 6247 CrossRef CAS PubMed;
(c) C. Zhang and M. L. Trudell, Tetrahedron Lett., 2000, 41, 595 CrossRef CAS.
-
(a) Z. Xiong, N. Wang, M. Dai, A. Li, J. Chen and Z. Yang, Org. Lett., 2004, 6, 3337 CrossRef CAS PubMed;
(b) S. M. Nobre and A. L. Monteiro, J. Mol. Catal. A: Chem., 2009, 313, 65 CrossRef CAS PubMed.
- D. Zim, S. M. Nobre and A. L. Monteiro, J. Mol. Catal. A: Chem., 2008, 287, 16 CrossRef CAS PubMed.
-
(a) S. U. Son, Y. Jang, K. Y. Yoon, E. Kang and T. Hyeon, Nano Lett., 2004, 4, 1147 CrossRef CAS;
(b) F. Lu, J. Ruiz and D. Astruc, Tetrahedron Lett., 2004, 45, 9443 CrossRef CAS PubMed;
(c) R. Tatumi, T. Akita and H. Fujihara, Chem. Commun., 2006, 3349 RSC.
-
(a) M. Cao, J. Lin, H. Yang and R. Cao, Chem. Commun., 2010, 46, 5088 RSC;
(b) V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588 CrossRef CAS PubMed;
(c) Y. Li, X. M. Hong, D. M. Collard and M. A. El-Sayed, Org. Lett., 2000, 2, 2385 CrossRef CAS PubMed.
- G. K. Rao, A. Kumar, B. Kumar and A. K. Singh, Dalton Trans., 2012, 41, 4306 RSC.
-
(a) F. Şen and G. Gökağaç, J. Phys. Chem. C, 2007, 111, 1467 CrossRef;
(b) S. E. Eklund and D. E. Cliffel, Langmuir, 2004, 20, 6012 CrossRef CAS;
(c) E. G. Castro, R. V. Salvatierra, W. H. Schreiner, M. M. Oliveira and A. J. G. Zarbin, Chem. Mater., 2010, 22, 360 CrossRef CAS;
(d) J. Alvarez, J. Liu, E. Roman and A. E. Kaifer, Chem. Commun., 2000, 1151 RSC;
(e) M. Cargnello, N. L. Wieder, P. Canton, T. Montini, G. Giambastiani, A. Benedetti, R. J. Gorte and P. Fornasiero, Chem. Mater., 2011, 23, 3961 CrossRef CAS;
(f) E. Sadeghmoghaddam, C. Lam, D. Choi and Y.-S. Shon, J. Mater. Chem., 2011, 21, 307 RSC;
(g) E. Sadeghmoghaddam, K. Gaïeb and Y.-S. Shon, Appl. Catal., A, 2011, 405, 137 CrossRef CAS PubMed;
(h) M. Moreno, L. N. Kissell, J. B. Jasinski and F. P. Zamborini, ACS Catal., 2012, 2, 2602 CrossRef CAS.
- M. Ganesan, R. G. Freemantle and S. O. Obare, Chem. Mater., 2007, 19, 3464 CrossRef CAS.
- J. D. Webb, S. MacQuarrie, K. McEleney and C. M. Crudden, J. Catal., 2007, 252, 97 CrossRef CAS PubMed.
-
(a) A. Kumar, G. K. Rao, S. Kumar and A. K. Singh, Dalton Trans., 2013, 42, 5200 RSC;
(b) Ar. Kumar, G. K. Rao, F. Saleem and A. K. Singh, Dalton Trans., 2012, 41, 11949 RSC;
(c) A. Kumar, G. K. Rao and A. K. Singh, RSC Adv., 2012, 2, 12552 RSC.
- G. K. Rao, A. Kumar, S. Kumar, U. B. Dupare and A. K. Singh, Organometallics, 2013, 32, 2452 CrossRef CAS.
-
(a) J. Rebek and F. Gavina, J. Am. Chem. Soc., 1974, 96, 7112 CrossRef CAS;
(b) J. Rebek, D. Brown and S. Zimmerman, J. Am. Chem. Soc., 1975, 97, 454 CrossRef CAS.
- V. P. Ananikov and I. P. Beletskaya, Organometallics, 2012, 31, 1595 CrossRef CAS.
- M. P. Lorenzo, J. Phys. Chem. Lett., 2012, 3, 167 CrossRef.
- F. Saleem, G. K. Rao, P. Singh and A. K. Singh, Organometallics, 2013, 32, 387 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data of L1 and 1; SEM, EDX and powder XRD of NPs. See DOI: 10.1039/c5ra00441a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 




