
Conversion of chitin and N-acetyl-D-glucosamine into a N-containing furan derivative in ionic liquids†
Xi Chena,
Yi Liub,
Francesca M. Kerton *b and
Ning Yan*a
*b and
Ning Yan*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, 117585, Singapore. E-mail: ning.yan@nus.edu.sg
bDepartment of Chemistry, Memorial University of Newfoundland, St. John's, NL A1B 3X7, Canada. E-mail: fkerton@mun.ca
First published on 12th February 2015
Abstract
Direct conversion of chitin to 3-acetamido-5-acetylfuran (3A5AF) in a range of ionic liquids (ILs) has been systematically investigated. 10 ILs with different cations and anions were tested as the solvent and 25 additives were screened. The results revealed that the presence of Cl in the IL is essential. In addition to the solubility enhancement of chitin in Cl containing ILs, the Cl anion appeared to participate in the chitin reaction cycle in IL solvents. 3A5AF can be obtained in some ILs, such as [BMIm]Cl, without any additive. Significantly enhanced yields of 3A5AF were obtained in [BMIm]Cl using boric acid and hydrochloric acid (HCl) as additives at 180 °C, a lower temperature than using organic solvents (215 °C). Kinetic studies showed that the product formed very quickly within 10 min, with much higher initial rate than using organic solvents. Recovered chitin was characterized by X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR) and elemental analysis (EA). In an effort to improve the yield, extraction and distillation were attempted for both chitin and chitin monomer, N-acetyl-D-glucosamine (NAG). Further studies were performed on NAG to see if acidic ILs would lead to enhanced reactivity. However, these were less effective.
1. Introduction
With growing concerns of depleting natural resources around the world, recent research has become focused on renewable feedstocks,1–3 of which lignocellulosic biomass draws the most scientific effort.4–7 Limited attention has been paid to ocean-based biomass. Between 2000 to 2012, less than 2.5% of about 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 publications on renewable feedstocks published concentrated on marine feedstocks.8 Therefore, ocean-based biomass, which holds a substantial potential for chemical production, has been underutilized. Chitin, an example of oceanic biomass, is the world's second most abundant biomass next to cellulose.9–11 It makes up 20–25% of crustacean shells such as crabs and prawns, which are readily available as waste from fishing and food industries. The abundance, sustainability and easy manufacture make it remarkably advantageous to utilize chitin as a resource for the production of various value-added chemicals. Moreover, chitin is made up of N-acetyl-D-glucosamine (NAG) monomers, which is an amino sugar inherently containing nitrogen. This unique property of chitin potentially enables the direct production of nitrogen-containing (N-containing) compounds from chitin, which cannot be achieved from lignocellulosic biomass. Currently, the synthesis of N-containing compounds starting from ammonia synthesis is usually laborious and energetically expensive. Transformation of chitin and related aminocarbohydrates into certain N-containing chemicals, therefore, is highly desirable.
000 publications on renewable feedstocks published concentrated on marine feedstocks.8 Therefore, ocean-based biomass, which holds a substantial potential for chemical production, has been underutilized. Chitin, an example of oceanic biomass, is the world's second most abundant biomass next to cellulose.9–11 It makes up 20–25% of crustacean shells such as crabs and prawns, which are readily available as waste from fishing and food industries. The abundance, sustainability and easy manufacture make it remarkably advantageous to utilize chitin as a resource for the production of various value-added chemicals. Moreover, chitin is made up of N-acetyl-D-glucosamine (NAG) monomers, which is an amino sugar inherently containing nitrogen. This unique property of chitin potentially enables the direct production of nitrogen-containing (N-containing) compounds from chitin, which cannot be achieved from lignocellulosic biomass. Currently, the synthesis of N-containing compounds starting from ammonia synthesis is usually laborious and energetically expensive. Transformation of chitin and related aminocarbohydrates into certain N-containing chemicals, therefore, is highly desirable.
Unfortunately, little has been done to transform chitin into chemicals until very recently. Chitin was converted into 5-hydroxymethylfurfural (5-HMF) and levulinic acid (LA).12,13 Chitin liquefaction in ethylene glycol via acid catalysis into a mixture of amino sugars has also been reported.14 Transformation of NAG, the monomer of chitin, has proved to be more successful.15–18 For example, 3-acetamido-5-acetylfuran (3A5AF) can be produced through pyrolysis at high temperatures19 and microwave irradiation15,16 with 2% and 62% yields, respectively. Nevertheless, NAG has to be obtained through the hydrolysis of chitin catalyzed by enzymes20–24 or concentrated acids,25 which is often inefficient and/or environmental unfriendly. In this regard, direct formation of small molecule chemicals from chitin in a single step would be desirable.26 We recently demonstrated the direct conversion of chitin into 3A5AF (Scheme 1) with ca. 7% yield. Unfortunately, N-methyl-2-pyrrolidone (NMP), an organic, toxic solvent and a high reaction temperature (at 215 °C for two hours) were needed.27
 | ||
| Scheme 1 Reaction of chitin conversion into 3A5AF product. | ||
Consequently, our further effort has been directed towards switching the solvent system to more environmentally benign ones, and to enable the reaction under milder conditions. To this end, we evaluated the possibility of conducting the reaction in ionic liquids (ILs), which are non-volatile, recyclable and possess better capability for chitin dissolution. ILs, which are completely composed of ions and are liquid at or close to room temperature,28,29 are suitable alternatives as the reaction media. In particular, ILs have proven to be excellent reaction media for cellulose- and lignin-based biorefinery processes.30–32 Detailed studies on direct transformation of chitin in ILs has not been described before, but it is not unreasonable to speculate that ILs may be suitable solvents for chitin conversion,33–36 because of the structural similarity between chitin and cellulose and that certain ILs exhibit higher capacity for chitin dissolution than organic solvents.33 In this study, 10 ILs and 25 additives were tested for conversion of chitin into furan products, and the influences of different IL and additives will be discussed in detail below. Reaction conditions were optimized and the recovered solids after the reaction were characterized. Comparable yield of 3A5AF could be obtained under milder conditions in ILs than NMP, with the optimum temperature dropped from 215 to 180 °C and enhanced reaction rates. In an effort to enhance the product yield, extraction and distillation have been attempted. Nevertheless, the yield was not improved significantly. As a result, experiments were performed on further optimizing 3A5AF production from the monomer sugar. We, and others, have shown that acidic additives significantly improve the yields of furans produced from NAG and other hexoses. In particular, acidic ionic liquids have been widely studied for carbohydrate to furan conversion processes,31,37 but have not been used in the dehydration of amino sugars. We thought that using such ionic liquids (examples in Chart 1) would simplify the reaction system and mean that other additives, such as boric acid, would no longer be needed.
 | ||
| Chart 1 The chemical structures of the used protic ILs. | ||
2. Experimental
2.1 Materials and characterization
Chitin was purchased from Wako Pure Chemical Industry. Boric acid was purchased from Amresco. Sodium chloride (NaCl) was bought from Schedelco. Lithium chloride (LiCl) was purchased from Alfa Aesar. NAG, barium, cesium, iron(II) and tin(II), chloride salts (BaCl2, CsCl, FeCl2 and SnCl2, respectively) were purchased from Sigma Aldrich. Other chemicals such as aluminium chloride (AlCl3), calcium chloride (CaCl2), chromium chloride (CrCl3), cobalt chloride (CoCl2), and nickel chloride (NiCl2) were obtained from Sinopharm Chemical Reagent (SCR). [AMIm]Br, [AMIm]Cl, [BMIm]Ac, [BMIm]BF4, [BMIm]Cl, [BMIm]CF3SO3, [BMIm]NTf2, [BMIm]PF6, [EMIm]Cl and [HOEMIm]Cl were purchased from Lanzhou Institute of Chemical Physics. All the chemicals were used as received. [BMIm]Cl, [BMIm]HSO4 and [BMIm-SO3H]HSO4 were prepared according to literature procedures38–40 or purchased from Alfa Aesar or Sigma Aldrich.X-ray powder diffraction (XRD), Fourier transform infrared spectroscopy (FTIR) and elemental analysis (EA) have been used for the characterization of chitin and the recovered solids. XRD with Cu Kα radiation at 40 kV was conducted on a Bruker D8 Advanced Diffractometer. The equations for crystalline index (CI), d-spacing and lattice size calculation are provided in the ESI.† FTIR was performed using a Bio-Rad FTS-3500 ARX instrument. EA was conducted using an Elementar Vario Micro Cube. High-performance liquid chromatography (HPLC) was performed using an Agilent 1200 Series (Agilent Technologies, Germany) LC system, with an Agilent ZORBAX Eclipse carbon-18 column and a UV-vis detector. The mobile phase consisted of 83% water and 17% acetonitrile at a flow rate of 0.5 mL min−1 and analysis time of 15 min. 3A5AF has peak absorbance at 230 nm. 3A5AF standard was prepared according to literature methods;15 a calibration curve was then plotted and applied for quantification of 3A5AF. GC-MS data were obtained using an Agilent Technologies 7890 GC with 5975 MSD and quantification performed as described elsewhere.16 Chloride levels were measured by ICP-MS (Perkin Elmer Sciex Elan DRC II ICPMS) and water levels through Karl-Fisher titration (Mettler Toledo C30 Coulometric KF Titrator) for both purchased and prepared ILs.
2.2 General procedures
After the reaction (typically 1 h), the reactor was cooled down to room temperature by cooling water. Methanol (20 mL) was added into each tube, after which the tube was sealed and vigorously shaken for 3 min. A portion of the methanol solution (1 mL) was filtered over a PTFE syringe filter (0.2 μm pore size) before HPLC analysis.
To determine the conversion of chitin, the remaining solid was isolated by centrifuging the reaction mixture, and washed with water three times before drying in an oven at 70 °C for 12 h. The formula below is used to calculate the conversion:
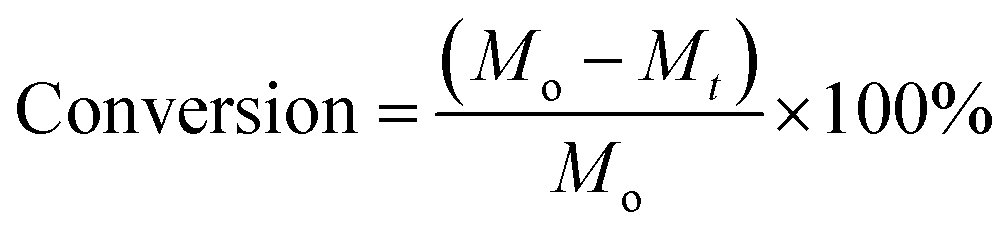
All reactions followed the general procedure, while varying a few parameters, namely solvent, additive, amount of additive, reaction time and temperature. In particular, for solvent screening, each ionic liquid (1 g) was added to each test-tube containing chitin (80 mg, 0.394 mmol), under three conditions: (1) without any additives; (2) with boric acid (97 mg, 1.57 mmol) only; and (3) with chromium chloride (105 mg, 0.394 mmol) only. Each sample was reacted at 180 °C for 1 h.
The recyclability of IL was examined in the following way. Chitin (80 mg, 0.394 mmol) with boric acid (97 mg, 1.57 mmol) were heated at 180 °C for 1 h in [BMIm]Cl (1 g). Then, 20 mL water was added and undissolved solids were removed by centrifugation. Next, the water phase was extracted by ethyl acetate (15 mL) for 3 times. Lastly, the water was evaporated under high vacuum at 90 °C. The recovered IL was reused by supplementing chitin and boric acid.
2.3 Solubility test
A simple solubility test of chitin in ionic liquids was conducted by mixing chitin (5 mg, 0.025 mmol) with various ionic liquids (0.5 g). After adding in a magnetic stirrer bar and sealing with Teflon caps, the mixtures were mixed at 100 °C for 1 h under stirring. Optical appearances of the final mixtures were recorded, to provide a rough comparison of solubility behavior of chitin in various ILs.2.4 Methods for the improvements of product yield
Extraction and distillation were attempted. The extraction was conducted by adding extra 3 mL of methyl isobutyl ketone (MIBK) into the reaction system as described in general procedures. Thus, the reaction system contains two layers with IL reaction layer at the bottom and MIBK extraction layer at the top. The installation for reactive distillation is shown in Scheme 2. The reaction was heated and reacted at 180 °C while a high-vacuum pump was applied during the reaction. An Edwards Rotary Vane Pump (Model RV3) with an ultimate pressure of 0.002 mbar was used. MIBK steam distillation was conducted by adding a heating unit to the reactive distillation system, which was the container and steam generator of water or MIBK by heating to the relative boiling point temperature.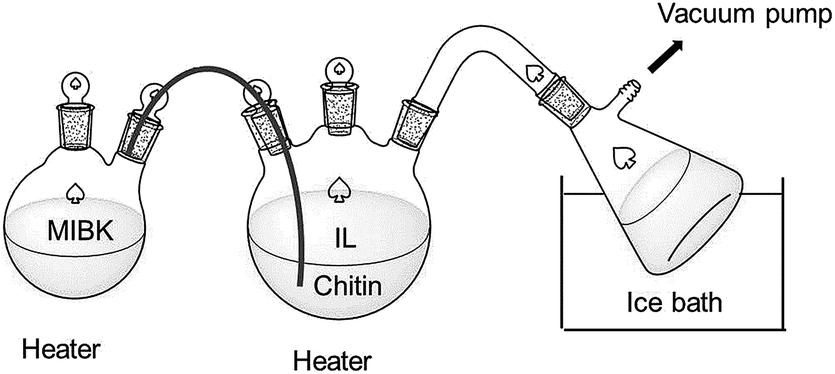 | ||
| Scheme 2 The diagram for reactive distillation process. | ||
3. Results and discussion
3.1 Solvent screening
10 different ILs with various cations and anions were used, including [AMIm]Br, [AMIm]Cl, [BMIm]Ac, [BMIm]BF4, [BMIm]Cl, [BMIm]CF3SO3, [BMIm]NTf2, [BMIm]PF6, [EMIm]Cl and [HOEMIm]Cl. Although several papers have described the dissolution behavior of chitin in ILs,33,41–43 contrary observations were reported possibly because the solubility not only depended on the structure of the ILs but also the sources of chitin, purity and so on. Thus, a simple solubility test (see Fig. 1) was conducted before the reaction. [AMIm]Br, [BMIm]Ac and [HOEMIm]Cl are able to dissolve chitin completely leading to clear solutions. Besides, [AMIm]Cl, [BMIm]Cl and [EMIm]Cl can partially dissolve chitin resulting in slightly opaque solutions. Nevertheless, [BMIm]PF6, [BMIm]BF4, [BMIm]NTf2 and [BMIm]CF3SO3 cannot dissolve chitin. Hydroxyl group functionalized IL, [HOEMIm]Cl, exhibited a better dissolution capability than [EMIm]Cl, possibly because the incorporation of a hydroxyl group increased the interaction between the solvent and the hydroxyl groups in chitin polymer chain. | ||
| Fig. 1 Solubility test of chitin in different ILs, (a) clear solution of chitin in [AMIm]Br, [BMIm]AC and [HOEMIm]Cl; (b) slightly opaque solution of chitin in [AMIm]Cl, [BMIm]Cl and [EMIm]Cl; (c) suspensions of chitin in [BMIm]PF6, [BMIm]BF4, [BMIm]NTf2 and [BMIm]CF3SO3. | ||
After the solubility test, blank experiments were conducted by adding chitin into the IL solvent at 180 °C for 1 h (see Fig. 2a). Surprisingly, 3A5AF was observed in [AMIm]Cl, [BMIm]Cl and [EMIm]Cl without any additive; whereas no 3A5AF was formed in pure organic solvents even at 225 °C.27 Note that all the three ILs in which 3A5AF was generated all contain Cl anion and exhibit partial solubility towards chitin. Nevertheless, [AMIm]Br, [BMIm]Ac and [HOEMIm]Cl which are able to completely dissolve chitin afforded no product, indicating that solubility is not the determining factor for the reaction, but Cl anion seems to be essential. To further substantiate our assumptions and screening the solvents, boric acid or CrCl3 were used as the additives, which have demonstrated effectiveness in cellulose/chitin conversion.21,44–46 The results were consolidated in Fig. 2b alongside the blank experiments. In all cases, only IL containing Cl anion formed 3A5AF products, which confirms the importance of Cl in these reactions. In the presence of additives, the yields improved remarkably. [BMIm]Cl and [EMIm]Cl showed the best performance with around 4.5% yield in the presence of boric acid and 2.3% yield with CrCl3. [HOEMIm]Cl produced 1.5 and 1.1% yield of 3A5AF in the presence of boric acid and CrCl3 respectively. However, in [AMIm]Cl, there was no product formation upon adding boric acid, and the yield in the presence of CrCl3 is comparable to the blank experiment. Overall, hydroxyl-functionalized cation-containing ILs were still less active in the presence of additives. Possibly the interaction between the functionalized cation and chitin chain inhibited the formation of 3A5AF. Based on these results, it was concluded that ILs with cations bearing alkyl chains and Cl as the anion were the most suitable solvents for chitin conversion into 3A5AF.
 | ||
| Fig. 2 (a) Blank experiments for chitin conversion in ILs; (b) chitin conversion in ILs with boric acid or CrCl3 additive. Reaction conditions: chitin (80 mg), IL (1 g), 400 mol% additive to substrate if added, 180 °C, 1 h. | ||
3.2 Additives screening
[BMIm]Cl was selected as the solvent in the following experiments because its performance was good and had the highest yield in blank experiments. For the screening of additives, single additives and combinations of additives were investigated. | ||
| Fig. 3 Comparison of yield of 3A5AF by using single or combinational additives. Reaction conditions: 180 °C, [BMIm]Cl (1 g), chitin (80 mg), single additive (100 mol%), with or without boric acid (400 mol%), 1 h. | ||
3.3 Optimization of reaction conditions for chitin conversion
The recyclability of [BMIm]Cl with boric acid as the promotor was examined. The IL was diluted with water after the reaction to remove the water insoluble parts. The organic compounds were removed by extraction. Finally, [BMIm]Cl was regenerated after removing water under high vacuum and reused with freshly added boric acid and chitin. The results were shown in Table S1.† A negligible decrease of yield was observed after the recycling of IL for 2 times. Although decomposition of [BMIm]Cl at 180 °C may occur,15 it was not severe and most of [BMIm]Cl still function in the recycling.
3.4 Characterization of recovered solids from chitin conversions
After the reactions, the unreacted chitin was recovered. XRD, FTIR and EA techniques were used for the characterization. The FTIR spectrum of recovered solid resembled that of pure chitin (see Fig. 4). In the FTIR spectra, the bands at 3487 and 3447 cm−1 represents the stretch of the OH, and those at 3261 and 3103 cm−1 represents the vibration of the NH.50 CH symmetric CH3 stretching and asymmetric CH2 stretching are represented by bands at 2889, 2932, 2960 cm−1, while the CH2 wagging, CH bending and CH3 symmetric deformation are attributed to peaks at 1313 and 1379 cm−1.51 The amide I band split into two peaks at 1661 and 1624 cm−1 indicating two types of inter-hydrogen bonds: the interaction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O with the N–H group and with the side chain group –CH2OH.52 Meanwhile, the amide II band is observed at 1555 cm−1. C–O bond stretching, asymmetric in-phase ring and asymmetric bridge oxygen stretching modes are represented respectively by the bands at 1028, 1072, 1117 and 1157 cm−1. Comparing the FTIR spectrum of recovered solid with that of pure chitin, it seems that most of the characteristic peaks remain unchanged in terms of the shape, position and relative size. It can thus be inferred that there were insignificant changes in the chemical structure such as functional groups, side chains and hydrogen bonding network after the reaction.
O with the N–H group and with the side chain group –CH2OH.52 Meanwhile, the amide II band is observed at 1555 cm−1. C–O bond stretching, asymmetric in-phase ring and asymmetric bridge oxygen stretching modes are represented respectively by the bands at 1028, 1072, 1117 and 1157 cm−1. Comparing the FTIR spectrum of recovered solid with that of pure chitin, it seems that most of the characteristic peaks remain unchanged in terms of the shape, position and relative size. It can thus be inferred that there were insignificant changes in the chemical structure such as functional groups, side chains and hydrogen bonding network after the reaction.
 | ||
| Fig. 4 FTIR spectra of chitin and recovered solid after reaction. | ||
XRD was employed to compare the crystallinity of the chitin before and after reaction. As shown in Fig. 5, the XRD spectra revealed no appreciable difference between pure chitin and recovered solid, as both have similar peak positions, intensities and shape. By using the XRD data, the CI, d-spacing and lattice size were calculated and are listed in Table 1. The CI value was almost maintained, indicating that the crystalline nature of chitin was not damaged during the course of the reaction. The lattice size became slightly bigger after the reaction than the initial pure chitin, which perhaps indicates a slight loosening of the inter-polymer chain interactions.
 | ||
| Fig. 5 XRD spectra of chitin and recovered solid after reaction. | ||
| Entry | CI/% | d spacing/nm | Lattice size/nm |
|---|---|---|---|
| Chitin | 91 | 0.466 | 9.7 |
| Recovered solid | 91 | 0.465 | 10.0 |
For composition analysis, EA was conducted and the results were shown in Table S1.† Consistent with the XRD and FTIR analysis, the difference in the composition before and after the reaction was not significant, indicating that the recovered solid largely maintained the chemical backbone of chitin. However, from the EA analysis, it can be seen that the nitrogen and especially the carbon content was higher in the recovered chitin. It was likely that carbonization happened and the recovered solid was a mixture of a small amount of carbonization product and unreacted chitin.53 To sum up, chitin retained its crystalline structure, hydrogen bonding network and chemical backbone after the reaction, but carbonization reaction may occur to a small extent.
3.5 Improvement of the product yield by extraction/distillation
The kinetic profile of this reaction was obtained by varying the reaction time from 3 to 120 min (see Fig. 6). Interestingly, the yield of 3A5AF reached 4.1% within 5 min and 5.2% within 10 min, respectively. Compared to the kinetic profile of chitin conversion in organic solvents, the dehydration rate has been accelerated in ILs and a much higher initial rate was been observed. However, after the initial 10 min, the yield increased quite slowly and reached a peak value if 6.2% at 60 min. By further prolonging the reaction time, the yield began to decrease slightly showing that the product may not be stable after 60 min. Therefore, although the use of ILs shows some advantages such as the more rapid reaction rate and lower temperature employed, the final yield of 3A5AF was not improved. According to the kinetic profile, it is reasonable to infer that possible product inhibition occurred during the reaction, which could explain the high initial reaction rate and much slower rate after 10 min. | ||
| Fig. 6 Kinetic studies of chitin dehydration into 3A5AF in IL and organic solvents. Reaction conditions in IL solvent: 180 °C, [BMIm]Cl (1 g), substrate (80 mg), boric acid (400 mol%), HCl (100 mol%); reaction conditions in organic solvent: 215 °C, NMP (3 mL), chitin (100 mg), boric acid (400 mol%), NaCl (200 mol%). | ||
As a result, extraction and distillation methods were considered for in situ product separation which may shift the equilibrium towards the production of 3A5AF and thus increase the yield. The extraction was achieved by simply adding MIBK solvent as an upper layer; this kind of reaction system has been reported previously and proved effective in 5-HMF extraction.54 However, no product was detected within the MIBK layer after the reaction and the yield was not enhanced in the presence of MIBK. This showed that the extraction of MIBK as an upper layer could not provide enough driving force to transfer the product from the IL layer. Afterwards, reactive distillation, which may have a stronger driving force, was employed to separate the 3A5AF product from the reaction system. Reactive distillation was attempted by (i) using a high-vacuum pump, and (ii) constantly passing the MIBK vapour through the IL layer, which have been successfully applied for 5-HMF separation and yield improvement by others.55 It is believed that by enabling the MIBK vapour to enter the IL layer offers more chances for drawing out the product than the two-phase extraction. Unfortunately, after several attempts, the product was not distilled out of the system. These two common methods for 5-HMF extraction and separation were not effective for 3A5AF separation in chitin conversion. Nevertheless, extraction of 3A5AF appeared to be effective if the starting material was N-acetyl-D-Glucosamine, see below.
3.6 N-Acetyl-D-glucosamine (NAG) conversion in protic ILs
Chitin monomer NAG was then used as the feedstock for further investigation due to its structural simplicity. A series of reactions using NAG were performed in order to (i) determine whether reactive extraction/distillation procedures show promise in the monomer conversion, and to (ii) confirm that hydrolysis of the chitin is one of the limiting steps in these reactions. Also, as addition of Brønsted acids (HCl, H3PO4) had shown synergistic effects when used with boric acid for conversion of chitin, we intended to determine whether protic ionic liquids such as [BMIm]HSO4 and [BMIm-SO3H]HSO4 were suitable media for these reactions and could be used in place of neutral ILs with aqueous acid additives.First, control reactions in [BMIm]Cl, [BMIm]HSO4 and [BMIm-SO3H]HSO4 were performed, where 100 mg NAG was dissolved in 750 mg [BMIm]Cl (or the corresponding molar equivalent of a protic IL) and heated to 180 °C for 3 min. Yields of 3A5AF were <1% from [BMIm]Cl, 7.8% from [BMIm]HSO4 and 0% from [BMIm-SO3H]HSO4. It should be noted that in our previous studies, a control reaction using [BMIm]Cl gave a yield of around 20% (ref. 15) and this was because the NAG used contained a boron-containing impurity that catalyzed the reaction.16 We postulated that the acidic reaction media might be entraining our product in the IL phase and therefore neutralizations were performed using a range of bases including NaHCO3. However, no significant differences in yield were observed. Next, extraction during reaction was attempted in [BMIm]Cl in the presence of two equivalents of boric acid. 3 mL MIBK was added into the vial as an extraction layer under microwave irradiation, and 30.2% yield of 3A5AF was obtained. Other solvents, which are useful solvents in the extraction of 5-HMF in biorefinery processes,56,57 including EtOAc, 1-butanol and 1-hexanol were also employed as the extraction layer, affording 3A5AF yield of 32.9%, 14.6% and 34.6% respectively. Considering the low boiling point and relatively environmentally-benign nature,58 EtOAc was selected as the most suitable solvent. When 3 mL EtOAc was present as an extraction solvent in [BMIm]HSO4, the yield increased moderately to 10.7% after 3 min, and further increased to 17.3% after 15 min. As [BMIm-SO3H]HSO4 did not produce any 3A5AF, and is more challenging to produce and thus more expensive than the other ILs studied, it was not investigated further. These results show that (i) the type of protic ionic liquid has a significant effect on the reaction outcome, (ii) lengthening reaction times can favourably increase yields, and (iii) the presence of a solvent in the microwave vial can serve to extract the product in situ and can lead to increased yields. The extraction method proves effective in NAG conversion under microwave irradiation, supporting our assumption that product inhibition might occur. However, such extraction was not successful in direct chitin conversion. This may be due to the increased viscosity and difficulty for the extraction from the chitin-IL system.
The effect of additives (boric acid and NaCl) on reactions in [BMIm]Cl and [BMIm]HSO4 in the presence of EtOAc, as an extraction layer, was studied. Addition of 1 molar equivalent or 2 molar equivalents of boric acid increased the yield in [BMIm]Cl from <1% to 19.1% and 32.9% respectively. Furthermore, if fresh EtOAc (3 mL) was added to the microwave vial of the latter experiment after the first EtOAc extraction layer had been removed and the mixture heated for another 3 min at 180 °C, an increase in yield to 40.4% was obtained. The yield was further increased to 48.8% by performing a third reactive extraction step. Increasing reaction time from 3 min to 9 min and increasing the volume of EtOAc used from 3 mL to 9 mL, the yield could be further increased to 55.6%. In contrast, reactions in the presence of two molar equivalents of boric acid in [BMIm]HSO4 had a decreased yield, 9.8% (cf. 10.7% with no boric acid). This shows that although HCl and other acids lead to synergistic effects with boric acid in the conversion of chitin to 3A5AF. There is no similar effect when NAG is used as the feedstock. This lends further credence to the hypothesis that the role of the mineral acid in the conversion of chitin is to hydrolyze the biopolymer. When NAG is used as the feedstock, there is no benefit in employing an additional acid and boric acid is superior at dehydrating the sugar to the desired furan. In agreement, with the studies on chitin described above, addition of one molar equivalent of NaCl (as a source of Cl ions) to a reaction in [BMIm]HSO4 led to an increased yield, 15.4% (cf. 10.7% without NaCl). It should be noted that for reactions involving added NaCl, the microwave temperature had to be ramped (1 min at 100 °C, 1 min at 120 °C, 1 min at 140 °C, 1 min at 160 °C and 1 min at 180 °C) to prevent overheating and temperature spikes because of the increased ion concentration and thereby increased conduction of microwave-heat. Temperature spikes could lead to rapid increases in vapor pressure within the microwave vials, which could lead to rupture of the vial. Addition of two equivalents of NaCl to the reaction in [BMIm]HSO4 gave a yield of 19.7%, which is close to double the yield in the absence of NaCl. These results confirm the importance of Cl ions in the formation of 3A5AF from both NAG and chitin.
4. Conclusion
In this paper, direct conversion of chitin to N-containing 3A5AF in a variety of ILs has been demonstrated. The solvent screening proved that the presence of Cl anion was a more significant factor than the solubility. Overall, the IL system appeared to be superior to the organic system. 3A5AF was formed in the blank and most of the Cl salts additive experiments by using IL as solvent; whereas no product was observed under the employed conditions by using organic solvent even at a higher temperature. Meanwhile, the kinetics showed that the product actually formed more quickly. By using [BMIm]Cl as the solvent and the combination of boric acid and HCl as the additives, comparable yield of 3A5AF could be obtained within 1 h at 180 °C. In an effort to enhance the yield, extraction and distillation were attempted. The strategies were not effective in chitin conversion whereas the extraction method was proved to be promising in NAG conversion. It suggests that improved methods for in situ chitin hydrolysis are needed, as a key step in its direct, one-pot transformation into N-chemicals. Therefore, IL functionalization aimed at assisting in this step may require more rational design. Future work may have to find other directions to improve the reaction yield from chitin which currently remain low.Acknowledgements
This work was supported by A*STAR, PSF funding with WBS number: R-279-000-403-305. FK thanks NSERC of Canada, Canada Foundation for Innovation, RDC-NL and Memorial University. She also thanks the RSC for a stopover in a commonwealth country travel grant.Notes and references
- J.-M. Park, A. Kondo, J.-S. Chang, C. Perry Chou and P. Monsan, Bioresour. Technol., 2013, 135, 1 CrossRef CAS PubMed.
- J. R. Rostrup-Nielsen, Science, 2005, 308, 1421–1422 CrossRef CAS PubMed.
- C. Somerville, H. Youngs, C. Taylor, S. C. Davis and S. P. Long, Science, 2010, 329, 790–792 CrossRef CAS PubMed.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- N. Yan and P. J. Dyson, Curr. Opin. Chem. Eng., 2013, 2, 178–183 CrossRef PubMed.
- X. Wang, L. Meng, F. Wu, Y. Jiang, L. Wang and X. Mu, Green Chem., 2012, 14, 758–765 RSC.
- F. M. Kerton, Y. Liu, K. W. Omari and K. Hawboldt, Green Chem., 2013, 15, 860–871 RSC.
- M. N. V. R. Kumar, React. Funct. Polym., 2000, 46, 1–27 CrossRef CAS.
- A. Domard, Carbohydr. Polym., 2011, 84, 696–703 CrossRef CAS PubMed.
- T. T. Franco and M. G. Peter, Polym. Int., 2011, 60, 873–874 CrossRef CAS.
- K. W. Omari, J. E. Besaw and F. M. Kerton, Green Chem., 2012, 14, 1480–1487 RSC.
- Y. Wang, C. M. Pedersen, T. Deng, Y. Qiao and X. Hou, Bioresour. Technol., 2013, 143, 384–390 CrossRef CAS PubMed.
- Y. Pierson, X. Chen, F. D. Bobbink, J. Zhang and N. Yan, ACS Sustainable Chem. Eng., 2014, 2, 2081–2089 CrossRef CAS.
- M. W. Drover, K. W. Omari, J. N. Murphy and F. M. Kerton, RSC Adv., 2012, 2, 4642–4644 RSC.
- K. W. Omari, L. Dodot and F. M. Kerton, ChemSusChem, 2012, 5, 1767–1772 CrossRef CAS PubMed.
- M. Osada, K. Kikuta, K. Yoshida, K. Totani, M. Ogata and T. Usui, Green Chem., 2013, 15, 2960–2966 RSC.
- F. D. Bobbink, J. Zhang, Y. Pierson, X. Chen and N. Yan, Green Chem., 2015, 17, 1024–1031 RSC.
- J. Chen, M. Wang and C.-T. Ho, J. Agric. Food Chem., 1998, 46, 3207–3209 CrossRef CAS.
- V. Choudhary, S. H. Mushrif, C. Ho, A. Anderko, V. Nikolakis, N. S. Marinkovic, A. I. Frenkel, S. I. Sandler and D. G. Vlachos, J. Am. Chem. Soc., 2013, 135, 3997–4006 CrossRef CAS PubMed.
- S. Dutta, S. De, M. I. Alam, M. M. Abu-Omar and B. Saha, J. Catal., 2012, 288, 8–15 CrossRef CAS PubMed.
- H. K. No, S. P. Meyers and K. S. Lee, J. Agric. Food Chem., 1989, 37, 575–579 CrossRef CAS.
- H. Sashiwa, S. Fujishima, N. Yamano, N. Kawasaki, A. Nakayama, E. Muraki and S.-I. Aiba, Chem. Lett., 2001, 308–309 CrossRef CAS.
- Y. Zhang, E. A. Pidko and E. J. M. Hensen, Chem.–Eur. J., 2011, 17, 5281–5288 CrossRef CAS PubMed.
- A. Einbu and K. M. Varum, Biomacromolecules, 2008, 9, 1870–1875 CrossRef CAS PubMed.
- X. Chen and N. Yan, Catal. Surv. Asia, 2014, 1–13 Search PubMed.
- X. Chen, S. L. Chew, F. M. Kerton and N. Yan, Green Chem., 2014, 16, 2204–2212 RSC.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- F. van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef CAS PubMed.
- P. Wang, H. Yu, S. Zhan and S. Wang, Bioresour. Technol., 2011, 102, 4179–4183 CrossRef CAS PubMed.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Chem. Rev., 2011, 111, 397–417 CrossRef CAS PubMed.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- W.-T. Wang, J. Zhu, X.-L. Wang, Y. Huang and Y.-Z. Wang, J. Macromol. Sci., Phys., 2010, 49, 528–541 CrossRef CAS.
- H. B. Xie, S. B. Zhang and S. H. Li, Green Chem., 2006, 8, 630–633 RSC.
- Y. Qin, X. Lu, N. Sun and R. D. Rogers, Green Chem., 2010, 12, 968–971 RSC.
- Q. Chen, A. Xu, Z. Li, J. Wang and S. Zhang, Green Chem., 2011, 13, 3446–3452 RSC.
- Z. Ding, J. Shi, J. Xiao, W. Gu, C. Zheng and H. Wang, Carbohydr. Polym., 2012, 90, 792–798 CrossRef CAS PubMed.
- L. Cammarata, S. Kazarian, P. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192–5200 RSC.
- S. Saravanamurugan, O. Nguyen Van Buu and A. Riisager, ChemSusChem, 2011, 4, 723–726 CrossRef CAS PubMed.
- X. Wang, W. Wu, G. Tu and K. Jiang, Zhongshan Daxue Xuebao, Ziran Kexueban, 2009, 48, 69–72 CAS.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Energy Fuels, 2010, 24, 737–745 CrossRef CAS.
- A. M. Bochek, A. A. Murav'ev, N. P. Novoselov, M. Zaborski, N. M. Zabivalova, V. A. Petrova, E. N. Vlasova, B. Z. Volchek and V. K. Lavrent'ev, Russ. J. Appl. Chem., 2012, 85, 1718–1725 CrossRef CAS.
- Y. Wu, T. Sasaki, S. Irie and K. Sakurai, Polymer, 2008, 49, 2321–2327 CrossRef CAS PubMed.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS PubMed.
- T. S. Hansen, J. Mielby and A. Riisager, Green Chem., 2011, 13, 109–114 RSC.
- T. Ståhlberg, S. Rodriguez-Rodriguez, P. Fristrup and A. Riisager, Chem.–Eur. J., 2011, 17, 1456–1464 CrossRef PubMed.
- Y. Su, H. M. Brown, X. Huang, X.-d. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361, 117–122 CrossRef CAS PubMed.
- Z. Zhang, Q. Wang, H. Xie, W. Liu and Z. Zhao, ChemSusChem, 2011, 4, 131–138 CrossRef CAS PubMed.
- A. Wang and T. Zhang, Acc. Chem. Res., 2013, 46, 1377–1386 CrossRef CAS PubMed.
- G. Cárdenas, G. Cabrera, E. Taboada and S. P. Miranda, J. Appl. Polym. Sci., 2004, 93, 1876–1885 CrossRef.
- F. G. Pearson, R. H. Marchessault and C. Y. Liang, J. Polym. Sci., 1960, 43, 101–116 CrossRef.
- M. Osada, C. Miura, Y. S. Nakagawa, M. Kaihara, M. Nikaido and K. Totani, Carbohydr. Polym., 2013, 92, 1573–1578 CrossRef CAS PubMed.
- P. Zhang, Y. Gong, Z. Wei, J. Wang, Z. Zhang, H. Li, S. Dai and Y. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 12515–12522 CAS.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- Z. Wei, Y. Liu, D. Thushara and Q. Ren, Green Chem., 2012, 14, 1220–1226 RSC.
- Y. Román-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef PubMed.
- F. Tao, H. Song and L. Chou, RSC Adv., 2011, 1, 672–676 RSC.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra00382b |
| This journal is © The Royal Society of Chemistry 2015 |