
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering a high energy surface of anatase TiO2 crystals towards enhanced performance for energy conversion and environmental applications
Wei
Chen
,
Qin
Kuang
*,
Qiuxiang
Wang
and
Zhaoxiong
Xie
*
State Key Laboratory of Physical Chemistry of Solid Surfaces, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, P.R. China. E-mail: qkuang@xmu.edu.cn; zxxie@xmu.edu.cn
First published on 5th February 2015
Abstract
Anatase titanium dioxide (A-TiO2) is one of the most important functional materials and is widely used in various energy- and environmental related applications. Over the past decade, great efforts have been devoted to surface engineering of A-TiO2 crystals at the atomic level so as to fundamentally understand the relationship between the surface structure and their performance in practical applications. In this review, we briefly summarize recent important achievements on the control of specific surface structures of A-TiO2 crystals, focusing on facets with high surface energy (such as {001}, {100}, {101}) and their combinations. In addition, fascinating performances of A-TiO2 crystals enhanced by these high energy surfaces are examined and discussed through the perspectives of synergistic effects of different facets and surface adsorbates, with additional insights related to some contradictory results. Finally, we offer a summary and some perspectives on current challenges and promising directions in this emerging field. We believe that a comprehensive understanding of surface engineering of A-TiO2 crystals with regard to high energy facets will in the long term help us to rationally design functional nanomaterials with desired performances.
1. Introduction
The surface is an important component of a solid state material, and different surfaces may exhibit different physical and chemical properties. This effect of the surface becomes especially prominent when the size of the solid is reduced to the nanoscale.1,2 For this reason, engineering surface structures, i.e. deliberately exposing specific facets with high energy and reactivity, is becoming a promising research direction in recent years and is conducted to improve the properties of the materials.3–13 However, high energy facets usually vanish in the bulk of crystals due to too fast a growth rate, and accordingly these thermodynamically stable facets preferentially predominate on the surface so as to minimize the total surface energy of crystals. It is a significant and challenging topic of nanomaterials to expose specific facets (especially high energy facets) on the surface of crystals.Titanium dioxide (TiO2) is one of the most studied functional nanomaterials because of its outstanding photoelectric and catalytic properties, as well as good structural stability, nontoxicity, low-cost and environmentally friendly nature.14 Among three common phases, anatase TiO2 (A-TiO2) attracts more attentions than rutile and brookite, as it presents better performances in various energy conversion and environmental applications, including photocatalytic and photoelectrocatalytic water splitting,15–17 air and water purification,18,19 reduction of CO2,20,21 photovoltaic cells,22–24 and lithium ion batteries (LIBs).25,26 To overcome some inherent limitations of A-TiO2 itself, such as low quantum efficiency and inactivation under visible light, various modification strategies, including doping foreign elements,27–29 constructing heterojunction,30–33 and loading sensitizers,34 have been developed in the past decades.35 However, these strategies seem to meet a bottleneck at present. Fortunately, recent breakthrough in metal nanocrystals gives us a hint that engineering high energy surface may be a more effective approach to enhance the performances of A-TiO2 crystals.9–11
For A-TiO2, the average surface energies of three fundamental low index facets follow the order of {001} (0.90 J m−2) > {100} (0.53 J m−2) > {101} (0.44 J m−2).36–38 Therefore the natural and artificial crystals of A-TiO2 are thermodynamically apt to expose the most stable {101} facets, which usually account for more than 90% of the total surface.36 In theory, the reactivity and activity of facets are proportional to their surface energy. For this, many efforts have been directed towards fabricating A-TiO2 crystals with exposed high energy and high reactivity facets.39–43 Excitingly, this strategy achieved great success in {001} facets.12,44–47 A large amount of studies have demonstrated that highly exposed {001} facets are favorable for enhancing performances of A-TiO2 crystals in photocatalysis,29,48,49 solar cells,50–52 and LIBs.53–56 Inspired by this success, controllable syntheses of other high energy facets, such as {100}, {110}, {111}, and {10l} (l > 1) facets are emerging out constantly within a short time.57–66 Noticeably, some research results contrary to conventional opinions have been reported.67–73 For example, Gordan et al. reported that the {101} facets of A-TiO2 were more reactive than the {001} facets for the photocatalytic H2 generation.70 And it was recently found that there was an optimal value of the relative ratio of {001} to {100} for enhancing photocatalytic performance.72 In addition, Pan et al. demonstrated that the clean facets followed the new photoactivity order of {010} > {101} > {001} in generating ˙OH radicals and hydrogen evolution, which does not accord with the order in their surface energy.73 These contradictory results make us confused but remind us that the surface energy may be not the only key factor to decide the performance of specific surface of A-TiO2 crystals, and we need to consider the surface-dependent performance at a deeper level. In recent years, there is more and more evidence that the synergistic effects between coexisting facets with different reactivities and the unpredictable effects of surface adsorbate species are responsible for confusing information.74–77 Unfortunately, the above two key issues are often ignored in present studies, especially when most of attentions are paid to those high activity facets.
In view of the above-mentioned facts, a timely review of surface engineering of A-TiO2 crystals and related performance enhancement seems necessary. Note that, even restricting to A-TiO2, it would be hard to provide an exhaustive overview of all the available works on this field. In this review, we only make a summary on recent important achievements on engineering surface structures of A-TiO2 crystals, with focus on those facets with high surface energy. In addition to {001} that are mostly concerned, {100}, {111}, {10l} (l > 1) facets and their combinations are equally discussed. Furthermore, enhanced performances of A-TiO2 crystals in energy conversion and environmental applications due to those high energy surfaces are examined, and the contradictory results are in particular discussed through the perspectives of synergistic effects of different facets and the surface adsorbates. We believe that this brief but deliberate review could be helpful for promoting the knowledge and understanding on this field.
2. The thermodynamically stable morphology of A-TiO2 crystals by predominately exposed {101} facets and related properties
Among all facets of A-TiO2, {101} facets are thermodynamically the most stable due to the lowest surface energy. According to the Wulff construction, the equilibrium shape of single A-TiO2 crystal is a slightly truncated tetragonal bipyramid enclosed by eight {101} facts and two {001} facets, where {101} facets on the side surface account for ca. 94% of the total surface area.36,37 The calculated shape of A-TiO2 crystal agrees well with the shape of naturally grown mineral sample, as shown in Fig. 1a and b.12,78,79 According to atomic structures of clean anatase {101} surface, the surface exposes both fivefold (Ti5C) and sixfold (Ti6C) coordinated Ti atoms, as well as twofold (O2C) and threefold (O3C) coordinated O atoms (Fig. 1c).79 In contrast, {001} facets as the minor surface of A-TiO2 crystals expose coordinatively unsaturated Ti5c and O2C atoms, as well as fully coordinated O3C (Fig. 1c). Such great difference in two surface atomic structures is bound to cause their distinct reactivity and properties. | ||
| Fig. 1 (a) The truncated tetragonal bipyramidal crystal form, showing the {001} and {101} facets. (b) An anatase crystal with a more usual aspect ratio and much larger {101} facets. Reprinted with permission from ref. 37. Copyright 2003 Elsevier B.V. (c and d) Atomic structures of unrelaxed, clean (101) and (001) surfaces. Reprinted with permission from ref. 79. Copyright 2008 Nature Publishing Group. | ||
As the most common facet exposed to the surface of A-TiO2, {101} facets have been investigated solely or as counterpart of {001} high energy facets in lots of theoretical and experimental studies.68,80–89 Due to higher stability, lower surface energy and less active sites, {101} facets are usually quite unreactive, displaying much poorer performance than {001} facets. For example, Amano et al. reported that A-TiO2 nanocrystals with a Wulff construction showed low H2 generation rate from aqueous methanol.67 They thought that the poor photocatalytic activity was closely associated with the surface structure of {101} facets, which would affect the reaction mechanism at the molecular level, rather than the conduction band level. According to the calculation results, chemisorption properties of A-TiO2 strongly depend on the surface structures of crystals, and water molecules are chemically adsorbed on {001} facets and physically adsorbed on {101} facets.78,90–93 Compared with the dissociative chemisorption of water molecules on {001} facets, the {101} facets is less favorable for dissociative adsorption of water and methanol, resulting in molecular nondissociative adsorption on them. In addition, the orientation-dependent charge-transfer process of A-TiO2 was experimentally investigated by electrochemical measurements on single crystal with exposed {001} and {101} facets.90,94 The results from Hengerer et al. revealed that water reduction, photo-oxidation and lithium insertion were favored on {001}, rather than {101}. These electrochemically orientational effects of A-TiO2 were ascribed to the differences in flatband potentials and surface atomic structures of {101} and {001} facets.90
On the other hand, it was likewise found in the study of Amano et al. that the performance of A-TiO2 octahedral crystals in photocatalytic decomposition of organic compounds under aerated conditions was more excellent than that of {001} faceted crystals.67 This exception was associated with the oxygenated environment and low density of defects on the well-crystallized surface, both of which could decrease the recombination of photogenerated carriers. Surprisingly, the selectivity for photocatalytic conversion of glycerol to hydroxyacetaldehyde in aqueous solution over {101} facets was superior to that over {001} facets, although the conversion of them were nearly the same.95 Very recently, theoretical calculations based on density-functional theory (DFT) has predicted that the A-TiO2 nanocrystals with predominately exposed {101} facets may be a promising candidate to be a component of the anode material in benthic microbial fuel cells due to the selective adsorption ability of the {101} surface to specific functional groups of biomolecules.96
Of note, in despite of the superiority in some applications, the inactive nature of {101} is fundamentally hard to overcome by conventional methods like doping and sensitizing. For this reason, increasing efforts have been recently devoted to designedly exposing those active facets with high surface energy so as to enhance the performance of A-TiO2.
3. Engineering high energy surface of A-TiO2 crystals by predominately exposed a single kind of specific high energy facets towards enhanced performances
3.1 {001} high energy facets
As mentioned above, {001} usually appears on the surface of truncated tetragonal bipyramidal A-TiO2 crystal as minor facets, coexisting with predominant {101} facets. Structurally, all Ti and O atoms on {001} are coordinatively unsaturated and the Ti–O–Ti bond angle is very large, which means that 2p states on the surface oxygen atoms are destabilized and very reactive.79 A large number of theoretical and experimental studies have proven that A-TiO2 crystals with higher percentage of exposed {001} facets show better activity than that with fewer {001} facet.7,45,58,97–99 For a long time, how to expose {001} facet with high percentage to the surface of micro- and nanocrystals is a great challenge for A-TiO2. In 2008, Yang and co-workers made a breakthrough in the controllable synthesis of A-TiO2 crystals.12 They successfully synthesized A-TiO2 microcrystals with 47% of exposed surface dominated by {001} facets, under the guidance of the prediction from the first-principle calculation. The key to success is the use of hydrofluoric acid (HF) that fluorates the surface of A-TiO2 crystals, whereby the surface energy of {001} is greatly reduced to a level below that of the original most stable {101}. The effectiveness of this synthetic strategy was quickly verified by subsequent successes.45,47,69,100–102 Strikingly, we promoted the percentage of exposed {001} facets to 89% by simply using high concentration of HF (∼47%) as the {001} facet capping agent.101 The bottom/top surface of as-prepared sheet-like A-TiO2 nanocrystals were enclosed by {001} facets and the average thickness was ca. 8 nm. As expected, such {001} faceted A-TiO2 nanosheets exhibited superior photocatalytic performance for the degradation of methyl orange (MO) to the commercially photocatalyst P25 (Fig. 2). More importantly, such performance advantages of {001} facets have been fully proven in both the photocatalytic oxidation reactions (such as degradation of pollutants) and photocatalytic reduction reactions (such as hydrogen production by water splitting).32,103–106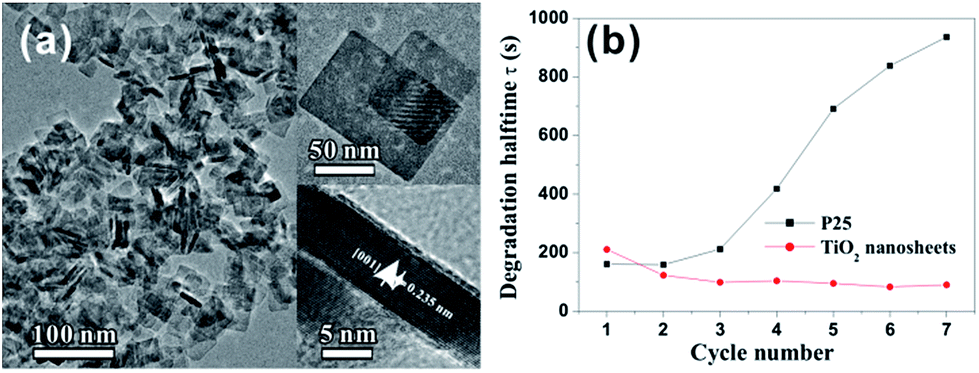 | ||
| Fig. 2 (a) High-magnification TEM image of TiO2 nanosheet; the insets show high-magnification TEM images of an individual nanosheet with different orientations. (b) Degradation half-life of MO for as-synthesized TiO2 nanosheets [89% (001) facets] and commercial P25 as a function of cycle number. Reprinted with permission from ref. 101. Copyright 2009 American Chemical Society. | ||
Note that the A-TiO2 nanosheets with highly exposed {001} facets also showed superior performance in other applications related to solar energy conversion, such as dye-sensitized solar cells (DSSCs).50,52,107–111 It has been proven that the higher percentage of the exposed {001} facet is, the higher overall conversion efficiency the A-TiO2 based DSSCs display (Fig. 3).107 The highest overall conversion efficiency reached 8.49% based on A-TiO2 with ca. 80% of exposed {001} facet, which was over 40% enhancement compared to the DSSCs based on Degussa P25. The excellent photovoltaic performance of A-TiO2 crystals with highly exposed {001} facet is attributed to the facts that the {001} facets not only possess superior light scattering and dye adsorption abilities but also can effectively retard the charge recombination due to high carriers transfer.52,107,109,111
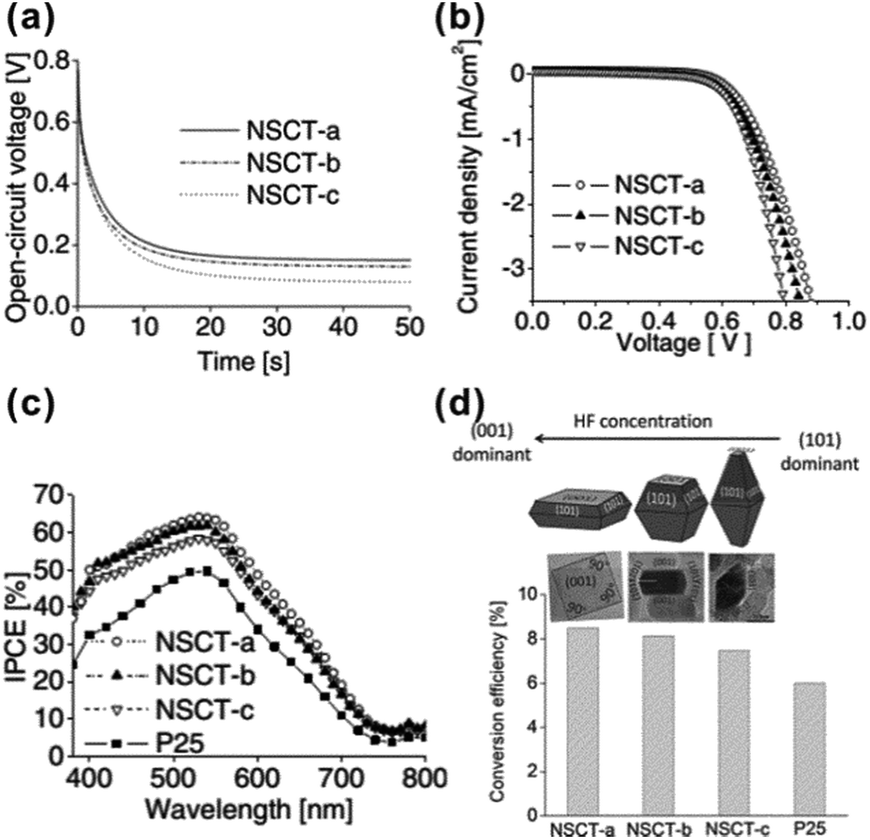 | ||
| Fig. 3 (a) Open-circuit voltage decay profiles, (b) dark current potential scans and (c) IPCE spectra of DSSCs based on the A-TiO2 crystals with different percentages of {001} (i.e., NSCT-a, NSCT-b, NSCT-c) and P25. (d) Schematic of the correlation between the particle morphology and the photovoltaic conversion efficiency of the A-TiO2 crystals with different percentages of {001} facets. Reprinted with permission from ref. 107. Copyright 2011 Wiley-VCH. | ||
Recent studies have further revealed that A-TiO2 nanocrystals with exposed {001} facets potentially exhibit an enhanced Li-ion insertion/extraction kinetics, including better reversibility and excellent rate capabilities.54,56,90,112 As we known, A-TiO2 has long been intensively studied as a promising electrode material for LIBs due to open channels in its structure that facilitates the lithium insertion/extraction during discharge/charge, while keeping the stability of the crystal framework. However, the lithium insertion/extraction kinetics in the crystal framework of A-TiO2 strongly depends on the orientation of A-TiO2 and {001} facets are more permeable for Li+ ions than {101} facets.90 This is because the diffusion of Li+ ions in the A-TiO2 framework occurs along a reaction path connecting the vacant octahedral interstitial sites, which makes diffusion more efficient along the [001] direction (the c axis direction) than in the plane normal to [001] direction. Furthermore, the energy barriers for Li-ion insertion through {001} and {101} facets of A-TiO2 (1.33 eV and 2.73 eV, respectively) are much higher than that for the bulk diffusion (0.35–0.65 eV).53,90,113 This indicates that the surface insertion is indeed the rate-determining step, and the transport of Li-ion is much faster across {001} facets than {101} facets. For this reason, ultrathin A-TiO2 nanosheets with highly exposed {001} facets are ideal electrode materials for LIBS due to the extremely short transport length scales in the [001] direction (Fig. 4).53,54,114 The success in A-TiO2 opens a new direction for improving the performance of electrode materials in LIBs by engineering their surface orientation.
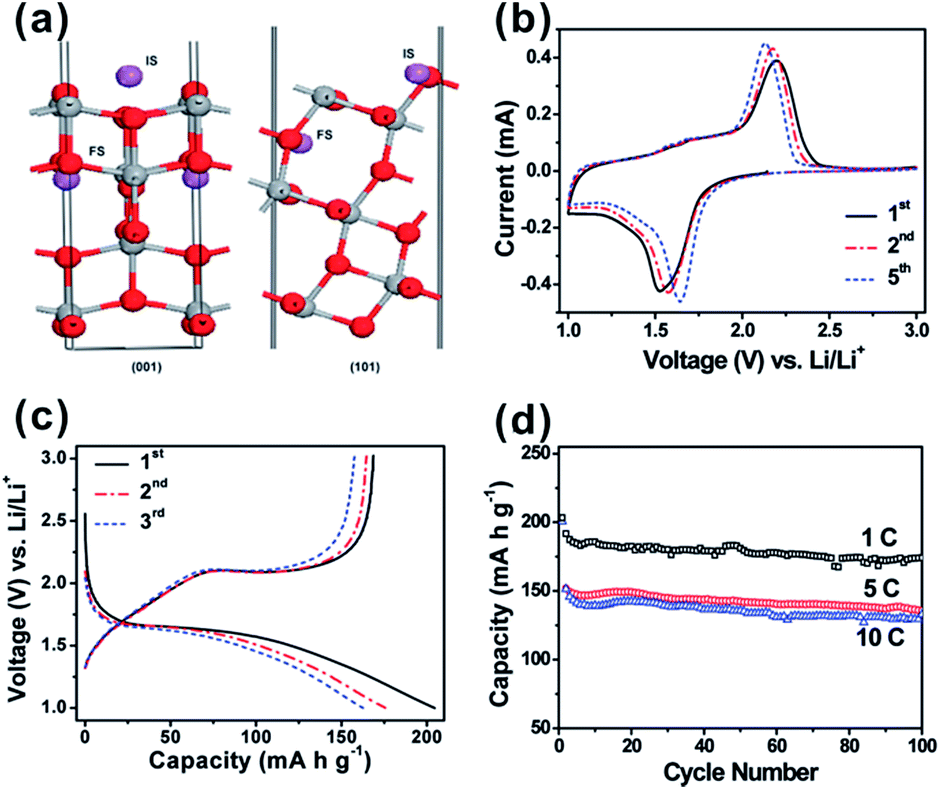 | ||
| Fig. 4 (a) Scheme of the anatase lattice with insertion of Li ions (purple spheres) between octahedral voids. The initial and final states of Li+ are indicated as ‘IS’ and ‘FS’, respectively. Grey spheres: titanium, red spheres: oxygen. Reprinted with permission from ref. 53. Copyright 2010 Royal Society of Chemistry. (b) Representative CVs at a scan rate of 0.2 mV s−1 for the first, second, and fifth cycles. (c) Charge–discharge profiles at a current rate of 5 C (850 mA g−1) for the first, second, and fifth cycles. (d) Cycling performance at different C rates. All of the measurements were conducted using a voltage window of 1.0–3.0 V. Reprinted with permission from ref. 54. Copyright 2010 American Chemical Society. | ||
It should be pointed out that the performances of A-TiO2 nanocrystals like nanosheets in applications are sometimes hampered by the face-to-face assembly of A-TiO2 nanocrystals and subsequent surface fusion due to hydrolysis of Ti–F groups on the {001} facets, which remarkably decreases the effective area of high active {001} facets.115,116 To avoid this, diverse hierarchical structures of A-TiO2, such as flowerlike microspheres and hollow boxes, are designedly fabricated by one-pot hydrothermal or solvothermal methods.49,52,54,103,108–110,114,117–120 Due to their hierarchical structures, these A-TiO2 crystals are endowed with some particular advantages, such as easy recyclability and superior light scattering effect, while keeping large effective area of {001} facets. Given that, the hierarchical A-TiO2 structures usually show superior performances in photocatalysis49,103,117,119,120 and DSSCs,52,108–110 to those unassembled samples.
In the synthetic processes mentioned above, HF and other fluorides are usually used as specific capping agent for {001} facets. Noticeably, the real role of HF is closely associated with synthetic conditions of A-TiO2 crystals, including concentration, solvent composition, reaction temperature and time. Under certain conditions, HF may play a role of chemical etching agent, rather than the desirable capping agent.118,121,122 Recent studies have revealed that in the presence of low-concentration of HF the available adsorption sites of A-TiO2 are occupied by F atoms to form the fluorinated surface; nevertheless, in the presence of high concentration of HF the completely fluorinated surface would be further dissolved to create surface vacancies.122 Interestingly, such a surface etching process merely occurs on the {001} facets, not on the {101} facets, due to the differences between the atomic arrangements on {001} and {101} facets. This is a revelation to us that some unusual facets of A-TiO2 may be designedly produced by means of rationally utilizing the dual roles of HF.
3.2 {100} high energy facets
As shown in Fig. 5a, the outermost Ti atoms of anatase {100} facets are fivefold coordinated, and the sixfold coordinated Ti (Ti6c) atoms belonging to the second layer lie at the bottom of grooves along the [010] direction.36 In contrast to the commonly exposed {001} and {101} facets, {100} facets are seldom presented on the surface of A-TiO2. As we know, the critical factor for controlling exposed facets of crystals is tuning the relative stability of different facets during the growth process, which is intrinsically determined by the average surface energy of the facets. The surface energy of {100} facets (0.53 J m−2) is slightly higher than that of {101} facets (0.44 J m−2), but much lower than that of {001} (0.90 J m−2).36–38 It is reasonable to predict that the {100} facets can be plentifully exposed by appropriately tuning growth conditions of A-TiO2 crystals. Fortunately, some research groups have got success in this field.57,58,62,63,73,123–126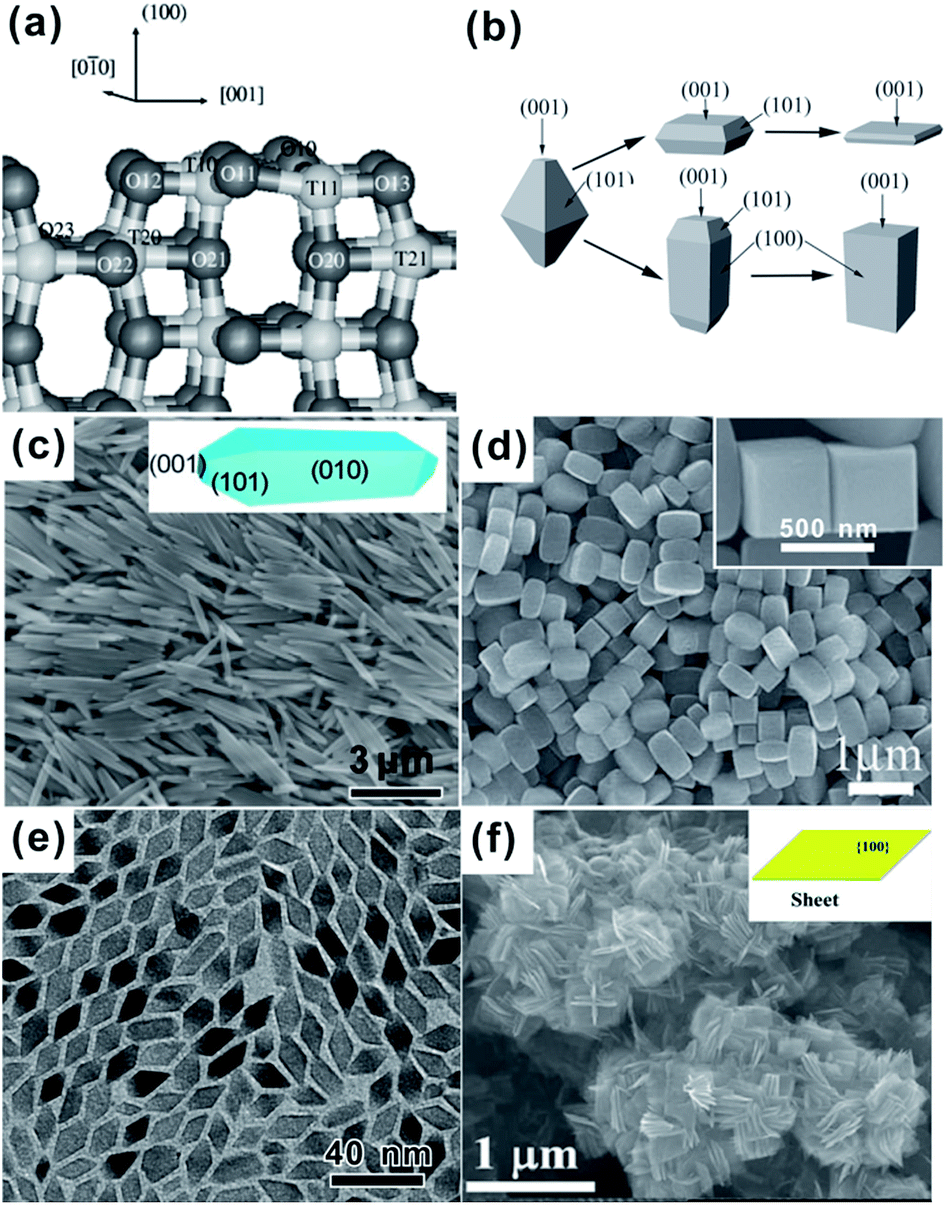 | ||
| Fig. 5 (a) The structure of the relaxed stoichiometric {100} facet. Reprinted with permission from ref. 36. Copyright 2001 American Physical Society. (b) Schematic drawings of typical morphologies of A-TiO2 with predominately {100} facets. Reprinted with permission from ref. 57. Copyright 2011 Wiley-VCH. (c) {100} faceted A-TiO2 rods. Reprinted with permission from ref. 63. Copyright 2011 Royal Society of Chemistry. (d) {100} faceted A-TiO2 cuboids (reprinted with permission from ref. 57. Copyright 2011 Wiley-VCH), (e) {100} faceted A-TiO2 rhombuses. Reprinted with permission from ref. 58. Copyright 2008 American Chemical Society, (f) {100} faceted A-TiO2 sheets. Reprinted with permission from ref. 123. Copyright 2013 American Chemical Society. | ||
As shown in Fig. 5b–f, A-TiO2 crystals with predominantly exposed {100} facets display as diverse forms, including rods,62,63,127–129 cuboids,57,60,73,130,131 rhombuses,58 and sheets.123 Rod-like, cuboidal, and sheet-like A-TiO2 crystals possess a tetragonal cross section, where the four side surfaces are bound by {100} facets. In either case, however, {100} facets always coexist with {001} and/or {101}, and the percentage of {100} in the total surface varies from 40% to 95%, depending on the aspect ratios of nanorods or the thickness of nanosheets.57,58,123,127 Note that {100} dominantly faceted A-TiO2 crystals are mostly prepared through the transformation of salt titanate precursors in basic solution under hydrothermal conditions.62,63,128–130 Barnard et al. have proposed by theoretical calculation that the surface energy of {100} facets can be lowered by surface hydroxyl groups, and thus the {100} facets are more stable than {101} and {001} facets in basic conditions.132,133 This mechanism pretty explains the formation of elongated truncated tetragonal bipyramids like rods or cuboids in basic solution, in which {100} facets are dominant and {101} and {001} only accounts for small percentages of the total surface (Fig. 5c and d).
Recent studies revealed that A-TiO2 crystals with dominantly exposed {100} facets can be also synthesized in the mixed solution of ionic liquid–water (1-butyl-3-methylimidazolium tetrafluoroborate, [bmim][BF4]),57 acidic solution containing HF,134–136 and even nonaqueous reaction system where the reaction medium is consisting of benzyl alcohol and oleic acid (or oleylamine) (Fig. 5e and f).58,123 In the mixed solution of ionic liquid–water, the ionic liquid [bmim][BF4] provided [bmim]+ and F− ions to stabilize {100} and {001} facets, respectively.57 DFT calculation indicates that the order of surface energy of {101}, {100}, and {001} facets can be reversed after surface fluorination, following the order of F-{001} < F-{100} < F-{101} (Fig. 6),135 and HCl as capping agent at (100) facets is so weakly bounded that it may be easily replaced by HF.134 This means that under acidic conditions, the exposed percentage of {100} facets can be delicately controlled by finely tuning the relative ratio of HF to HCl. However, the mechanism for the formation of {100} faceted A-TiO2 rhombic crystals in nonaqueous reaction system has no exact answer at present, but a trace amount of water in situ formed in organic solvent is supposed to be responsible for that.58
 | ||
| Fig. 6 DFT calculated surface energies (the unit is J m−2) and structures for different stages of HF interaction with single crystal anatase TiO2 (101) (left) (001) (middle) and (100) (right) surfaces. (a) Clean surfaces; (b) full HF-covered surfaces; (c) complete fluorinated surfaces. All structures are optimized structures. Reprinted with permission from ref. 135. Copyright 2012 Royal Society of Chemistry. | ||
Among the three basic facets, {001} facets with the highest surface energy are usually considered to possess the best performance. However, it has been recently reported that {100} facets could show much better performances in photocatalytic H2 generation and CO2 reduction than {001} and {101} facets due to the higher conduction band minimum and more oxygen vacancies on {100} facets.57,73,123 Besides, enhanced photoelectric conversion efficiency, depending on the exposed percentage of {100} facets, was also obtained on the thin-film electrodes made from A-TiO2 crystals exposed high percentage {100} facets.63,131 It is well-accepted that the distinctive surface structure of {100} facets such as the superior electron structure, gifts them some excellent properties including higher conduction band minimum, more Ti5c atoms and oxygen vacancies and so on, in comparison with {001} and {101} facets.41,63,69,73,123 These characteristic properties should account for the excellent performances shown on {100} facets.
3.3 High index facets of [010] zone axis
In addition to the widely studied facets mentioned above, the controlled exposure of other facets with high surface energy, such as high index facets of [010] zone axis, such as {401} and {10l} (e.g., {102}, {103}, {105}, and {106}) high index facets, is sporadically reported in previous studies.61,64–66,137,138 The high index facets of [010] zone axis ({10l} (l > 1) and {h01} facets) are high index facets that can be described as a combination of {101} and {001} (or {100}) basic crystal facets as the terraces and steps in different modes, as shown in Fig. 7a. | ||
| Fig. 7 (a) Schematic model of stepwise {101}, {102}, {103}, and {401} surfaces projected along the [010] direction. SEM images, TEM images, and ideal models of TiO2 particles with exposed (b1–b3) {102}, (c1–c3) {103} and (d1–d3) {401} facets. Reprinted with permission from ref. 64 and 66. Copyright 2012 Wiley-VCH and 2013 Wiley-VCH. | ||
Despite there is great difficulty in syntheses, {10l} and {401} facets can be predominantly exposed on the surface of A-TiO2 and even form a fully faceted polyhedron.64,66 {102} and {103} facets are the early reported {10l} high index facets, which were first synthesized through transformation of potassium titanate nanowires with the assistance of hexamethylenetetramine under solvothermal conditions.64 As shown in Fig. 7b and c, {102} facets constitute a pudgy tetragonal bipyramidal A-TiO2 with the interfacial angles of 76° between the opposite facets near the tips, while {103} and {101} facets coexist on the surface of A-TiO2 to form a sixteen-faceted polyhedron where eight equivalent {103} facets account for 60% of the total surface and the interfacial angles between the opposite facets near the tips is 101°. In contrast, {401} faceted A-TiO2 crystals present a spindly octahedral morphology with the interfacial angles of 11° near the tips, which is much smaller than that of the pudgy tetragonal bipyramids enclosed with {102} (Fig. 7d).66 It should be stated that, the topotactic structural transformation from the solid titanate precursor to A-TiO2 is the key to achieve exposing these high index facets. This transformation process is essentially a solid reaction, and thus the reaction rate is much slower compared to previous synthetic routes based on the direct hydrolysis of titanium precursors. Because of this, the capping agents are endowed unique opportunity to finely tune growth rates of various facets by specific adsorption on the stepped sites of high index facets.
Besides the solution syntheses, {10l} faceted A-TiO2 crystals can be synthesized through a high-temperature gas-phase oxidation route. For example, pudgy tetragonal bipyramidal A-TiO2 nanocrystals with eight equivalent {105} facets were successfully synthesized.65 It was observed in their time-dependent experiments that the formation of the {105} faceted tetragonal bipyramidal A-TiO2 undergo a two-stage growth process from the truncated tetragonal bipyramid with {101} and {001} facets to the intermediate crystals enclosed by {101} and {107} facets. Such special {105} faceted A-TiO2 microcrystals are regarded as a result of the synergistic effect of thermodynamic and kinetic factors that control the crystal nucleation and subsequent epitaxial growth on the existing {001} facet during the vapor deposition.
Different from the {10l} cases mentioned above, {10l} facets can be dominantly exposed to the surface along with {100} facets. For example, by tuning the concentration of F− ions in a hydrothermal reaction,138 {106} and {100} facets constitute rectangular A-TiO2 nanosheets, where {106} facets appear as the top/bottom surface and account for 56% of the total surface. The introduced F− ions play a key role in exposing {106} facets, which triggers the distinctive olation for the formation of this special crystal facets.
Structurally, {10l} facets should be highly active, since they possess high-density atomic steps and unsaturated coordinated sites. According to DFT calculation, the surface energy of {105} facets is lower than that of {001}, but much higher than that of {101}.65 And theoretical and experimental studies demonstrated that the {105} facets possessed the capability to cleave water photocatalytically to generate hydrogen. Besides that, the A-TiO2 crystals with exposed {102} and {103} facets have proven to present more superiority than A-TiO2 octahedron with only exposed {101} facets for the degradation of methylene blue (MB). The order of photodegradation efficiencies for different facets was concluded as {001} > {102} ≈ {103} > {101}.64 Surprisingly, recent study revealed that {401} faceted A-TiO2 nanocrystals exhibited better electrochemical performance in LIBs than truncated tetragonal bipyramids with highly exposed {001} facets.66 It is a pity that more systematic investigation of the structure–performance relationship with regard to these high index facets is seriously subject to the difficulty in syntheses.
3.4 Other higher energy facets
Among the low index facets of A-TiO2, both {110} (1.09 J m−2) and {111} (1.61 J m−2) facets possess far higher surface energy than all the basic facets discussed above (i.e., {101}, {100} and {001}).36,38,139 Consequently, they cannot be dominated on the surface of A-TiO2, but usually appear as the third or fourth party on the surface of A-TiO2, along with dominant {101} and {001} facets.59,61,139,140 Liu et al. first reported that the {110} high energy facets can be artificially exposed by using Ti power as precursor and HF and hydrogen peroxide (H2O2) as facet capping agents.61 Four {110} facets were emerged as rhombus on the surface of tetragonal pyramidal A-TiO2 crystals with dominantly exposed {001} and {101} facets, where {110} facets account for merely a few percentage. In this synthetic process, H2O2 was considered the decisive factor for exposing {110} facets to the surface of A-TiO2. It was proposed that the Ti4+ precursor can react with H2O2 to form yellow peroxotitanium acid (Ti2O5(OH)x(x−2)−, x = 1–6), which retards the hydrolysis rate of the titanium precursor and provides enough time for F− ions to tune the growth behavior by selectively adsorption. In fact, this formation mechanism mediated with a solid transformation is very similar with the cases of {10l} high index facets. Structurally, the {001} facets are composed of 100% Ti5C atoms, while the {110} facets are composed of 50% Ti4C atoms and 50% Ti6C atoms. Due to the existence of steric effects, F− ions adsorbs easier to Ti5C than to Ti4C. Consequently, the percentage of {110} facets could be elevated to 11% by optimizing the proportions of H2O2 and HF in reaction solution.140Among the reported facets, {111} facets present the highest surface energy (1.61 J m−2). So far only one report is known to successfully expose {111} facets to the surface of A-TiO2 crystals.139 As shown in Fig. 8a and b, {111} appears as rhombic facets that are formed by truncating eight corners of elongated tetragonal bipyramidal crystals originally dominated with {101} {100} and {001} facets. The high percentage emergence of {111} could be ascribed to the synergetic effect of the F− and ammonia as the capping reagents. This kind of A-TiO2 crystals showed an enhanced photocatalytic activity in photocatalytic water splitting compared to other samples with predominantly exposed {100}, {101}, and {001} (Fig. 8c). This is ascribed to the reason that the conduction and minimum of {111} is much higher than those of {100}, {101}, and {001} (Fig. 8d). It should be pointed out that {110} and {111} facets usually appear as the third party or fourth on the surface of A-TiO2, and they only account for a very small percentage of the surface. For this reason, it is hard to directly identify the effects of {110} or {111} on the performance of A-TiO2.
 | ||
| Fig. 8 (a) Structure model of A-TiO2 and (b) morphology model of A-TiO2 crystals with exposed {111} facets. (c) Photocatalytic water splitting tests of the Pt-loaded (0.5%) TiO2 samples dominantly exposed with {111} (T010), {100} (T010), {101} (T101), and {001} (T001) facets, respectively. (d) Schematic illustration of the determined valence band (VB) and conduction-band (CB) edges of T001, T101, T010, and T111. Reprinted with permission from ref. 139. Copyright 2013 American Chemical Society. | ||
4. Two key issues in engineering high energy surface of A-TiO2 crystals towards enhanced performances
It is clearly seen from Section 3 that exposing high-energy facets to the surface, in most cases, can effectively enhance their performance in practical applications. However, there are also considerable contradictory results in the {001} and {100} cases. The main reasons leading to the above issues may arise from two aspects: one is the synergism of a low percentage of those co-existing facets that is often ignored when our attentions are focused on those so-called active facets, and the other is the synergistic effect of surface adsorbates that are still unclear to us. In the following sections, we will make particular discussions on the key issues.4.1 The synergistic effects of coexisting facets on the photocatalytic performance of A-TiO2
Except few cases of {10l} faceted octahedron, A-TiO2 crystals are always enclosed by two or more groups of facets, even for A-TiO2 nanosheets with predominately exposed a specific group of facets. When we pay attentions to those high energy facets, the thermodynamically stable facets like {101} cannot be ignored. Because of the anisotropy of crystals, different facets have distinct properties, such as adsorption and coordination capacity as well as reaction activity. A rational use of these differences by engineering surface structure may trigger synergistic effects between different facets on the surface of crystals with coexisting facets, thereby further enhancing the performance of the crystals.72,141,142 Nevertheless, the facet matching, i.e. the simultaneous exposing of right facets is the precondition for positively influencing the photocatalytic performance of A-TiO2 crystals. As counter-example, the transfer of photogenerated electrons and holes would go along the same direction in the A-TiO2 with coexisting {001} and {100} facets, which may greatly inhibit the photocatalytic activity of A-TiO2.142 Next, we will explicate the synergistic effects between multiple facets of TiO2 on their photocatalytic performance through a few typical examples.As the most common morphology, truncated tetragonal bipyramidal A-TiO2 crystals with coexisting {001} and {101} facets have been widely studied. As a matter of course, the synergistic effect between {001} and {101} facets for the photocatalytic performance is firstly concerned by researchers. Because A-TiO2 crystals predominately expose {101} facets under natural growth, the control of different ratios of {001} to {101} facets can be regarded as the controllable exposure of {001} facets. As a result, the higher percentage exposure of high-energy {001} facets is usually considered as the primary reason for the enhanced photocatalytic performance.45,46,104 In fact, the enhanced performance of these anatase with specific surface structures depended not only on the high percentage exposure of {001} facets, but also on some other factors, especially the spatial separation of redox sites on the different crystal facets.69,74,104,141,143
In 2002, Matsumura et al. investigated the positions of redox sites on the surface of A-TiO2 by photo-activated selective deposition.77 Under irradiation, Pt and PbO2 particles were deposited on {101} and {001} facets, respectively, which indicates that the {101} and {001} facets provide reduction and oxidation sites in the photochemical reaction, respectively. Recently, increasingly developed in situ characterization techniques have provided us more intuitive evidences with regard to the spatial separation of redox sites on {001} and {101} facets of A-TiO2.75,76,144 As shown in Fig. 9a, it can be in situ observed by a single-molecule fluorescence approach on single particle that the redox-responsive fluorogenic dyes are preferentially reduced on {101} facets, which indicates that the effective reduction sites are located on the {101} facets of the crystal rather than the {001} facets.75,144 In addition, the electron spin resonance (ESR) spectra, which directly relates the reactivity of different facets with the type, the amount, and the location of electronic defects, likewise verified that the oxidation sites (O− centers) and the reduction sites (Ti3+ centers) locate on {001} and {101} facets, respectively (Fig. 9b).76 The essential reason for spatial separation of redox sites is that the slight difference of the local energy band structures between {101} and {001} facets drives the directional separation of photogenerated electrons and holes to these facets, respectively.77,145 According to DFT calculation,141,146,147 the Fermi level of {001} facets enter their valence band, while the Fermi level of {101} facets is still located at the top of the valence band of {101} surface, as shown in Fig. 10a. When both {001} and {101} facets are exposed to the surface of A-TiO2 crystals, the different Fermi levels of anatase {001} and {101} facets induce the position variance of energy bands of them. Consequently, a so-called “surface heterojunction” could be formed between {001} and {101} on the surface of A-TiO2, which is functionally similar with the known phase-heterojunctions built by two semiconductors with different energy band structures.148–151 The construction of surface heterojunction makes it possible that photogenerated electrons and holes will preferentially transfer to {101} and {001} facets, respectively, thereby leading to different reactivity on these facets (Fig. 10b).146 Given the efficient spatial separation of photogenerated carriers, the co-exposure of {101} and {001} is obviously the right combination, which are beneficial for enhancing the photocatalytic performance of A-TiO2 crystals.
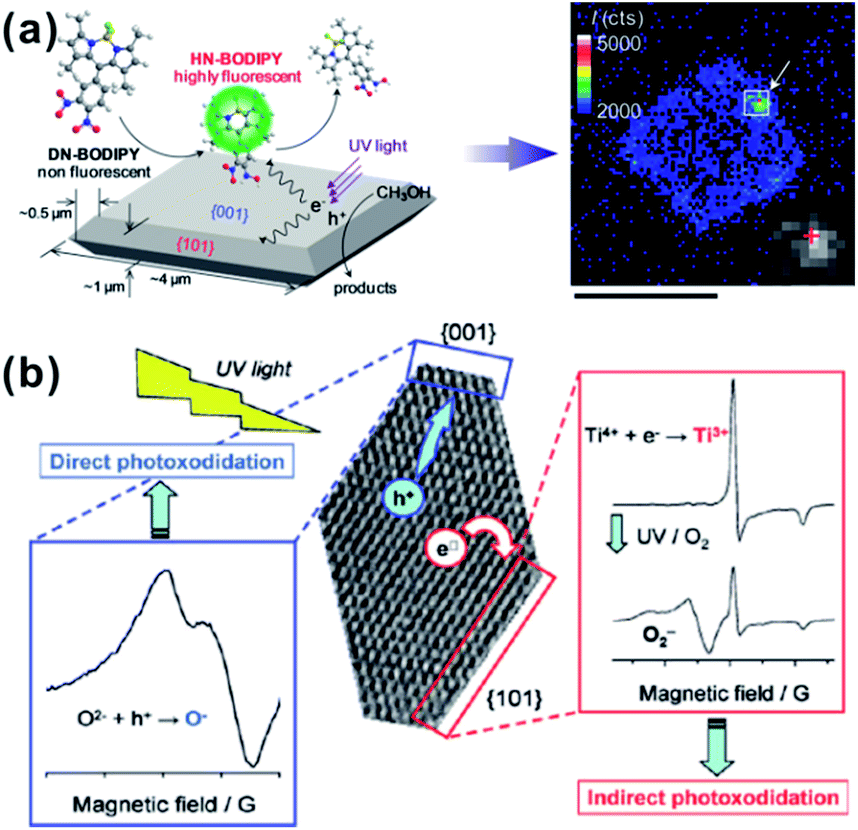 | ||
| Fig. 9 (a) Schematic of in situ observation photocatalytic reduction of single fluorescent molecule (HN-BODIPY) from nonfluorescent molecule (DN-BODIPY) over a TiO2 crystal. Reprinted with permission from ref. 75. Copyright 2011 American Chemical Society. (b) Schematic of experimental ESR spectra on different facets of truncated tetragonal bipyramidal TiO2 nanocrystals. Reprinted with permission from ref. 76. Copyright 2011 American Chemical Society. | ||
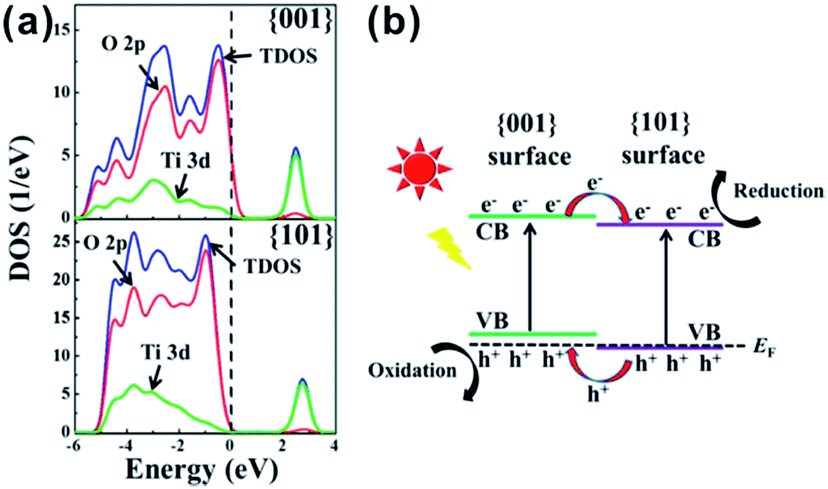 | ||
| Fig. 10 (a) Density of states (DOS) plots for {101} and {001} surface of A-TiO2. (b) {001} and {101} surface heterojunction. Reprinted with permission from ref. 146. Copyright 2014 American Chemical Society. | ||
A photocatalytic process is known to involve two half reactions, i.e. photo-reduction and photo-oxidation, which are associated with the photogenerated electrons and holes, respectively. Since {101} and {001} facets exhibit the reduction and oxidation reactivity in the photocatalytic process, respectively, an appropriate relative ratio of {001} to {101} facets is bound to play the key role in the cooperative processing of the photo-reduction and photo-oxidation half reactions. Of note, the optimal ratio of {001} to {101} facets varies from case to case, depending on synthetic methods of A-TiO2 crystals and types of measured photocatalytic processes.48,72,74,141,146,152 For example, the surface-fluorinated A-TiO2 nanocrystals with exposed 70% {001} facets was reported to have the highest performance in degradation of acetone,48 but in H2 evolution, the highest efficiency occurred over the surface-fluorinated sample with 45% {001} facets.141 In addition, for the fluorine-free A-TiO2 nanocrystals synthesized by using carbonate ions as capping agent, the optimal percentage of {001} facts is ca. 60% for degradation of methylene blue.152 This indicates that the optimal ratio in different cases is likewise influenced by the adsorbed species on the crystal surface. Because of a strong capping effect for {001}, the fluorine species are difficult to be completely removed from the surface of A-TiO2 crystals, and thus the surface-fluorinated degree would influence the photocatalytic performance of photocatalysts to some extent.32,48,100,143 In this regard, it is better to obtain A-TiO2 crystals with clean surface for investigating the real photocatalytic performance, although the synergistic effect of coexisting {001} and {101} facets has been confirmed.
As we discussed above, the functionally matching of coexisting facets is the precondition for enhancing the photocatalytic performance of A-TiO2 crystals. In contrast to the right combination of {101}/{001}, it is questionable whether there is the synergistic effect of {100}/{001} facets. In the study reported by Zhao et al., anatase cuboids with exposed about 80% {100} facets and 20% {001} exhibited superior photocatalytic activity than sheet-like TiO2 with exposed dominant {001} and minor {101}.57 However, some contrary results were also obtained by Zan group.124,129,142 They intentionally synthesized two similar sheet-like crystals with the coexistence of {101}/{001} and {100}/{001}, respectively. By comparing the generation of hydroxyl radicals (˙OH) and superoxide radicals (O2−), they found that the photogenerated charge carriers were efficiently separated and photocatalytic oxidation and reduction respectively took place on {001} and {101} facets over the crystals. While {101} facets were replaced by {010} facets, the photocatalytic activity was inhibited (Fig. 11a and b).142 On the basis of photo-activated deposition of Ag nanoparticles on the two samples, they proposed that the transfer of photogenerated electrons and holes would go along the same direction in the A-TiO2 with coexisting {001} and {100} facets, which greatly inhibits the photocatalytic activity of A-TiO2 (Fig. 11c–f). In addition, the reconstruction of different facets under UV light irradiation changed the situation of unsaturated coordinated Ti atoms on each kind of facets, which leads to the lower photocatalytic activity of {100} facets than that of {001} and {101} facets.124 On the other hand, they also confirmed the superior photocatalytic activity of {100} facets for the photoreduction of CO2 to CH4 than that of {001} facets in spite of the reversed performances were exhibited after Pt-loading.129
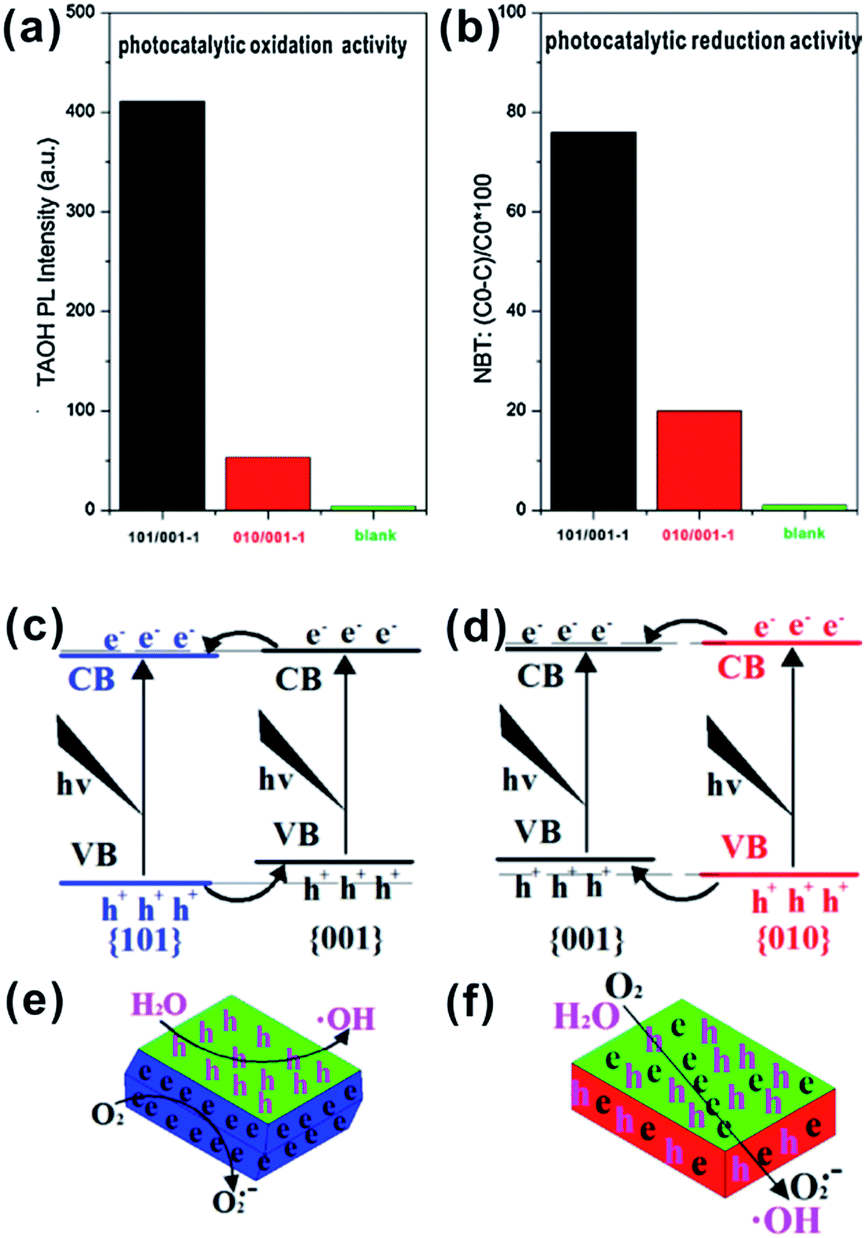 | ||
| Fig. 11 Comparison of photocatalytic oxidation (a) and reduction (b) activity of 101/001−1 and 010/001−1. (c) Electronic band structures of {101}–{001}; (d) electronic band structures of {010}–{001}; (e) electrons and holes distributing of {101}–{001}; and (f) electrons and holes distributing of {010}–{001}. Reprinted with permission from ref. 142. Copyright 2013 Elsevier B.V. | ||
It should be stated that, solely engineering surface structure by optimizing exposed facets is rather limited in enhancing the overall photocatalytic performance of A-TiO2 crystals, although the separation of photogenerated carriers can be significantly enhanced by tuning the relative ratio of {001} to {101}.72 Most of photocatalysts are wide bandgap semiconductors, and need to cooperate with other metallic or semiconducting functional materials as co-catalysts, which facilitate photo-excited redox reactions by providing the active sites/reaction sites or strengthening the charge separation.153–157 For example, noble metals (especially Pt) as co-catalysts not only serve as electron sinks, suppressing the charge recombination, but also provide effective proton reduction sites, thereby dramatically improving photocatalytic efficiency of semiconductor photocatalysts.158 Given that, it is a great potential for acquiring highly efficient photocatalysts to rationally combine the facet-induced effect and heterojunction-induced effect with regard to photogenerated carriers.159 To achieve this, the selective deposition of co-catalysts, i.e. loading oxidation and/or reduction co-catalysts on the respective faces of semiconductor photocatalysts, is a feasible strategy. For example, by selectively depositing an appropriate amount of Pt nanoparticles on the {101} photo-reductive facets, the photocatalytic performances of A-TiO2 in the photocatalytic oxidation process (i.e., photodegradation of methyl orange, Fig. 12a) and the photocatalytic reduction process (i.e., H2 evolution from splitting water, Fig. 12b) were strikingly boosted.72 It has been demonstrated that this strategy is universal and applies to other semiconductor photocatalysts, such as BiVO4, WO3, AgI, BiOCl, and so on.159–162
 | ||
Fig. 12 (a) Degradation curves of MO in the presence of TiO2 truncated tetragonal bipyramidal nanocrystals without deposition of Pt (naked TiO2, —●—), with selective deposition of Pt on {101} facets (TiO2–Pt(S)-0.5%, —■—), and with nonselective deposition of Pt (TiO2–Pt(NS)-0.5%, —▲—), respectively. Inset shows the corresponding kinetic rate curves (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C/C0 ∼ t). (b) H2 evolution amounts in the presence of the above three photocatalysts after irradiation for 6 h. Reproduced with permission from ref. 72. Copyright 2013 Wiley-VCH. C/C0 ∼ t). (b) H2 evolution amounts in the presence of the above three photocatalysts after irradiation for 6 h. Reproduced with permission from ref. 72. Copyright 2013 Wiley-VCH. | ||
As of now, A-TiO2 with variously coexisting facets has been successfully synthesized. Unsatisfactorily, the prepared crystals have not been well studied and the synergistic effect and functional mechanism of the coexisting facets also has not been well elucidated, besides that the combination of {101} and {101}. Fortunately, the positive effect for enhancing the performance is conspicuous based on A-TiO2 that co-exposes suitably and ratio-optimally different facets. It encourages us to unremittingly explore and reasonably design A-TiO2 with coexistence of different facets to further improve the properties of them.
4.2 The effects of surface adsorbate species on the photocatalytic performance of A-TiO2
As we mentioned in Section 3 the capping agents, such as HF and other fluorides are one of the essential elements for controllable exposure of {001}, {100}, or other high energy facets. However, the facet-dependent performances of A-TiO2 crystals remain disputed, with the interference from surface adsorbed species (HF or F− ions). For example, a large number of studies have reported that residual fluoride species on the surface deteriorated the photocatalytic performance of A-TiO2 to a certain extent, due to the reduced surface energy.12,45,57,73,101,141 In contrast, a considerable number of studies have likewise showed that the fluorinated A-TiO2 sample exhibited better photocatalytic performance than the defluorinated sample.32,48,100,106,163–165 It is usually considered that the positively enhancement effect of the fluoride species adsorbed results from the strong O2-capturing ability of the![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Ti–F(H) group on the fluorinated surface, which can promote the transfer of photogenerated electrons and then retard the recombination of photogenerated electrons with photogenerated holes (Fig. 13).32,48,100,163 In addition, surface fluorination of A-TiO2, which may produce more Ti atoms with lower coordination numbers (e.g. Ti4c) through surface reconstruction, results in more favourable sites on A-TiO2 for reactants in photocatalysis.32
Ti–F(H) group on the fluorinated surface, which can promote the transfer of photogenerated electrons and then retard the recombination of photogenerated electrons with photogenerated holes (Fig. 13).32,48,100,163 In addition, surface fluorination of A-TiO2, which may produce more Ti atoms with lower coordination numbers (e.g. Ti4c) through surface reconstruction, results in more favourable sites on A-TiO2 for reactants in photocatalysis.32
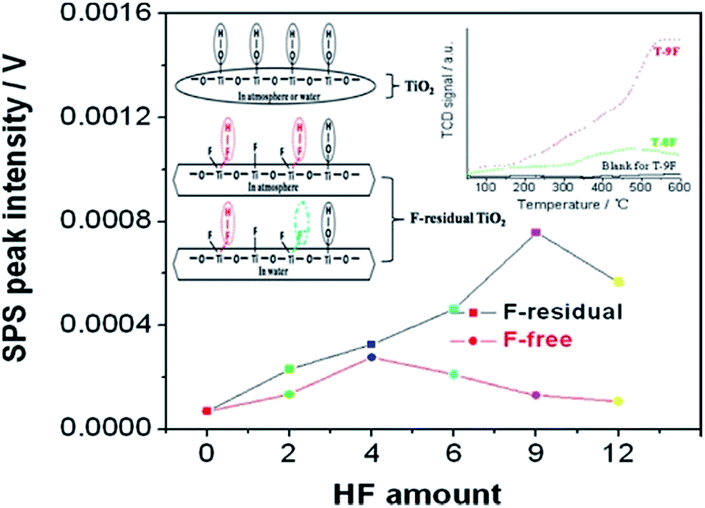 | ||
| Fig. 13 Intensities of SPS peak at 350 nm of resulting F-residual TiO2 and its corresponding F-free one. Insets are surface structure schematic of common TiO2 and the F-residual one in the atmosphere or in water. Reprinted with permission from ref. 163. Copyright 2013 American Chemical Society. | ||
Given that the confusing effects of the fluorinated surface, it is highly desirable to acquire A-TiO2 with clean surface, which is conducive to making clear the intrinsic surface structure–performance relationship of A-TiO2. The calcination under high temperature and the long-time ion exchange with high concentration of NaOH solution are usually adopted to clean up the fluorinated surface of A-TiO2 crystals.12,101,164 However, the net effects of the two treatment methods are often far from being satisfactory. For example, the high-temperature treatment is likely to cause a phase transition from anatase to rutile and the aggregation of nanocrystals, and also the surface fluorides are hard to be completely removed via ion exchange. In this regard, it is of more practical significance to develop alternative synthetic methods without HF for the preparation of A-TiO2 with exposed {001} facets.45,47,117,166–168 Carbonate ions are recently proven to be a kind of effective stabilizer of anatase {001} facets, because the surface energy of {001} facets with high intensity of unsaturated coordinated Ti4+ atoms can be remarkably reduced by bidentate-chelating with carbonate ions.64,152 Compared to HF, carbonate ions are easier to remove by heat treatment or acid treatment because of high chemical instability. In contrast to solution reactions mentioned above, the gas phase process based on chemical vapour deposition is better for preparing A-TiO2 with clean surface. Unfortunately, the facet-controllable synthesis is very difficult to achieve in gas phase process, and thus relevant works are rarely reported.65,169
5. Summary and outlook
With the rapid development of theoretical calculations and experimental methods, numerous studies have been obtained in the fields of engineering the surface structures of A-TiO2 and making clear their surface structure–performance relationship. In this review, we tried to summarize recent research progresses on the control of specific exposed facets of A-TiO2 crystals. The commonly exposed facets, such as {001}, {101}, {100}, and their combinations, have been widely studied and rich in the results of theories and experiments. Some high-energy or high-reactivity crystal facets rarely and even firstly observed, such as {10l}, {111} and {110}, are also mentioned. The facet-dependent performances in practical applications, especially in photocatalysis, are well interpreted through the perspectives of the surface atomic structures and local electronic structures. Besides, the cooperative mechanism between coexisting facets is another significant factor and potential for further improving the properties of A-TiO2 crystals. In fact, the synergistic effect of coexisting facets is derived from the so-called surface junction formed by adjacent facets, which efficiently promotes the separation of the photogenerated charge carriers. However, the real situation in practical applications would be far more complex than we discussed here. Each possible factor, such as the crystal size,69,86 morphology,170 adsorbed species,70 structural defects in surface and bulk,171 surface construction,37etc. could influence the performance of A-TiO2 crystals. Since the influence of each factor is not clear and interrelated, they should not be considered individually in fact.Although a great progress has been made to date, there are still many problems to be resolved in further research. Rationally engineering surface structure, i.e. exactly exposing specific facets, is always our first priority. Most of the present approaches to synthesize A-TiO2 with exposed specific facets are based on the capping effect of F-containing compounds, which are hazardous to humans and not easy to be removed. In order to obtain TiO2 crystals with clean surface that can exhibit real performance, new strategies with reduced or no fluorine species should be developed. Furthermore, considering TiO2 crystals are usually used as model system for studying many important surface properties and potentially used in practical application fields, the synthetic strategies should be also suitable for large scale production in high quality. To achieve this, both factors of thermodynamics and kinetics during the growth of nanocrystals must be taken into accounts, and the after-treatment such as selectively etching is likely a worthwhile strategy. Secondly, the surface adsorbed species inevitably cause some unexpected influences on the performance of A-TiO2 crystal. Thus, the real effects of adsorbed species at different situations will need to be uncovered. Thirdly, it may be of more significance to make the facet-induced effect effectively cooperate with other surface modification strategies, such as surface deposition by co-catalysts. The strategy of rationally cooperating the facet effect and heterojunction effect is currently restricted to the case of truncated bipyramidal A-TiO2 with {101} and {001}. However, we believe that this synergism strategy would be the most promising direction of acquiring high-efficient and low cost catalysts.
It is no doubt that opportunities and challenges coexist in the controllable syntheses of A-TiO2 with specific surface structures. As we know, TiO2 is strategic for cutting-edge areas of science and has been intensively investigated as model system bridging the material gap between surface science and practical applications in a long term. Therefore, the comprehension on the surface–performance relationship in energy- and environmental applications achieved from A-TiO2 can be reference to other inorganic functional nanomaterials and other application fields. If so, it will takes us one step closer to rationally designing functional nanomaterials with desired performances.
Acknowledgements
This work was supported by the National Basic Research Program of China (2011CBA00508 and 2015CB932301), the National Natural Science Foundation of China (21171142, 21131005, 21333008 and 21473146).Notes and references
- M. Haruta and M. Daté, Appl. Catal., A, 2001, 222, 427–437 CrossRef CAS.
- L. M. Molina and B. Hammer, Appl. Catal., A, 2005, 291, 21–31 CrossRef CAS PubMed.
- X. Xie, Y. Li, Z.-Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- Z.-Y. Jiang, Q. Kuang, Z.-X. Xie and L.-S. Zheng, Adv. Funct. Mater., 2010, 20, 3634–3645 CrossRef CAS PubMed.
- N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- Q. Kuang, X. Wang, Z. Jiang, Z. Xie and L. Zheng, Acc. Chem. Res., 2014, 47, 308–318 CrossRef CAS PubMed.
- X. Han, M. Jin, S. Xie, Q. Kuang, Z. Jiang, Y. Jiang, Z. Xie and L. Zheng, Angew. Chem., Int. Ed., 2009, 48, 9180–9183 CrossRef CAS PubMed.
- Y. Ma, Q. Kuang, Z. Jiang, Z. Xie, R. Huang and L. Zheng, Angew. Chem., Int. Ed., 2008, 47, 8901–8904 CrossRef CAS PubMed.
- Z.-Y. Zhou, N. Tian, J.-T. Li, I. Broadwell and S.-G. Sun, Chem. Soc. Rev., 2011, 40, 4167–4185 RSC.
- Z. Quan, Y. Wang and J. Fang, Acc. Chem. Res., 2012, 46, 191–202 CrossRef PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–642 CrossRef CAS PubMed.
- K. Zhou and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 602–613 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS PubMed.
- M. A. Henderson, Surf. Sci. Rep., 2011, 66, 185–297 CrossRef CAS PubMed.
- K. Shankar, J. I. Basham, N. K. Allam, O. K. Varghese, G. K. Mor, X. Feng, M. Paulose, J. A. Seabold, K.-S. Choi and C. A. Grimes, J. Phys. Chem. C, 2009, 113, 6327–6359 CAS.
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- O. M. Alfano, D. Bahnemann, A. E. Cassano, R. Dillert and R. Goslich, Catal. Today, 2000, 58, 199–230 CrossRef CAS.
- K. Mori, H. Yamashita and M. Anpo, RSC Adv., 2012, 2, 3165–3172 RSC.
- S. Das and W. M. A. W. Daud, RSC Adv., 2014, 4, 20856–20893 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- Z. S. Wang, H. Kawauchi, T. Kashima and H. Arakawa, Coord. Chem. Rev., 2004, 248, 1381–1389 CrossRef CAS PubMed.
- F. Zhu, D. Wu, Q. Li, H. Dong, J. Li, K. Jiang and D. Xu, RSC Adv., 2012, 2, 11629–11637 RSC.
- Z. Weng, H. Guo, X. Liu, S. Wu, K. W. K. Yeung and P. K. Chu, RSC Adv., 2013, 3, 24758–24775 RSC.
- A. G. Dylla, G. Henkelman and K. J. Stevenson, Acc. Chem. Res., 2013, 46, 1104–1112 CrossRef CAS PubMed.
- Q. Xiang, J. Yu, W. Wang and M. Jaroniec, Chem. Commun., 2011, 47, 6906–6908 RSC.
- G. Liu, H. G. Yang, X. Wang, L. Cheng, J. Pan, G. Q. Lu and H.-M. Cheng, J. Am. Chem. Soc., 2009, 131, 12868–12869 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, Phys. Chem. Chem. Phys., 2011, 13, 4853–4861 RSC.
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2011, 112, 1555–1614 CrossRef PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed.
- J. Yu, L. Qi and M. Jaroniec, J. Phys. Chem. B, 2010, 114, 13118–13125 CAS.
- L. Qi, J. Yu and M. Jaroniec, Phys. Chem. Chem. Phys., 2011, 13, 8915–8923 RSC.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- G. Liu, L. Wang, H. G. Yang, H.-M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 155409 CrossRef.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 119901 CrossRef.
- C. Z. Wen, H. B. Jiang, S. Z. Qiao, H. G. Yang and G. Q. Lu, J. Mater. Chem., 2011, 21, 7052–7061 RSC.
- W.-J. Ong, L.-L. Tan, S.-P. Chai, S.-T. Yong and A. R. Mohamed, ChemSusChem, 2014, 7, 690–719 CrossRef CAS PubMed.
- G. Liu, J. C. Yu, G. Q. Lu and H.-M. Cheng, Chem. Commun., 2011, 47, 6763–6783 RSC.
- C.-T. Dinh, T.-D. Nguyen, F. Kleitz and T.-O. Do, ACS Nano, 2009, 3, 3737–3743 CrossRef CAS PubMed.
- W. Q. Fang, X.-Q. Gong and H. G. Yang, J. Phys. Chem. Lett., 2011, 2, 725–734 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, S.-P. Chai, S.-T. Yong and A. R. Mohamed, Nanoscale, 2014, 6, 1946–2008 RSC.
- H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H. M. Cheng and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 4078–4083 CrossRef CAS PubMed.
- S. Liu, J. Yu and M. Jaroniec, Chem. Mater., 2011, 23, 4085–4093 CrossRef CAS.
- D. Zhang, G. Li, X. Yang and J. C. Yu, Chem. Commun., 2009, 4381–4383 RSC.
- Q. Xiang, K. Lv and J. Yu, Appl. Catal., B, 2010, 96, 557–564 CrossRef CAS PubMed.
- S. Liu, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2010, 132, 11914–11916 CrossRef CAS PubMed.
- J. Yu, J. Fan and K. Lv, Nanoscale, 2010, 2, 2144–2149 RSC.
- L. Etgar, P. Gao, Z. Xue, Q. Peng, A. K. Chandiran, B. Liu, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2012, 134, 17396–17399 CrossRef CAS PubMed.
- H. Zhang, Y. Han, X. Liu, P. Liu, H. Yu, S. Zhang, X. Yao and H. Zhao, Chem. Commun., 2010, 46, 8395–8397 RSC.
- C. H. Sun, X. H. Yang, J. S. Chen, Z. Li, X. W. Lou, C. Li, S. C. Smith, G. Q. Lu and H. G. Yang, Chem. Commun., 2010, 46, 6129–6131 RSC.
- J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah, D. Luan, S. Madhavi, F. Y. C. Boey, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 6124–6130 CrossRef CAS.
- J. S. Chen, L. A. Archer and X. W. Lou, J. Mater. Chem., 2011, 21, 9912–9924 RSC.
- J. S. Chen and X. W. Lou, Electrochem. Commun., 2009, 11, 2332–2335 CrossRef CAS PubMed.
- X. Zhao, W. Jin, J. Cai, J. Ye, Z. Li, Y. Ma, J. Xie and L. Qi, Adv. Funct. Mater., 2011, 21, 3554–3563 CrossRef CAS PubMed.
- B. Wu, C. Guo, N. Zheng, Z. Xie and G. D. Stucky, J. Am. Chem. Soc., 2008, 130, 17563–17567 CrossRef CAS PubMed.
- L. Pan, J.-J. Zou, S. Wang, X.-Y. Liu, X. Zhang and L. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 1650–1655 CAS.
- Y. Miao and J. Gao, Micro Nano Lett., 2011, 6, 848–851 CAS.
- M. Liu, L. Piao, L. Zhao, S. Ju, Z. Yan, T. He, C. Zhou and W. Wang, Chem. Commun., 2010, 46, 1664–1666 RSC.
- J. Li and D. Xu, Chem. Commun., 2010, 46, 2301–2303 RSC.
- J. Pan, X. Wu, L. Wang, G. Liu, G. Q. Lub and H.-M. Cheng, Chem. Commun., 2011, 47, 8361–8363 RSC.
- X. Han, B. Zheng, J. Ouyang, X. Wang, Q. Kuang, Y. Jiang, Z. Xie and L. Zheng, Chem.–Asian J., 2012, 7, 2538–2542 CrossRef CAS PubMed.
- H. B. Jiang, Q. Cuan, C. Z. Wen, J. Xing, D. Wu, X.-Q. Gong, C. Li and H. G. Yang, Angew. Chem., Int. Ed., 2011, 50, 3764–3768 CrossRef CAS PubMed.
- X. Han, F. Zhou, L. Li and C. Wang, Chem.–Asian J., 2013, 8, 1399–1403 CrossRef CAS PubMed.
- F. Amano, T. Yasumoto, O.-O. Prieto-Mahaney, S. Uchida, T. Shibayama and B. Ohtani, Chem. Commun., 2009, 2311–2313 RSC.
- N. Wu, J. Wang, D. N. Tafen, H. Wang, J.-G. Zheng, J. P. Lewis, X. Liu, S. S. Leonard and A. Manivannan, J. Am. Chem. Soc., 2010, 132, 6679–6685 CrossRef CAS PubMed.
- G. Liu, C. Sun, H. G. Yang, S. C. Smith, L. Wang, G. Q. Lu and H.-M. Cheng, Chem. Commun., 2010, 46, 755–757 RSC.
- T. R. Gordon, M. Cargnello, T. Paik, F. Mangolini, R. T. Weber, P. Fornasiero and C. B. Murray, J. Am. Chem. Soc., 2012, 134, 6751–6761 CrossRef CAS PubMed.
- S. Kalluri, K. H. Seng, Z. Guo, H. K. Liu and S. X. Dou, RSC Adv., 2013, 3, 25576–25601 RSC.
- C. Liu, X. Han, S. Xie, Q. Kuang, X. Wang, M. Jin, Z. Xie and L. Zheng, Chem.–Asian J., 2013, 8, 282–289 CrossRef CAS PubMed.
- J. Pan, G. Liu, G. Q. Lu and H.-M. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133–2137 CrossRef CAS PubMed.
- N. Murakami, Y. Kurihara, T. Tsubota and T. Ohno, J. Phys. Chem. B, 2009, 113, 3062–3069 CAS.
- T. Tachikawa, S. Yamashita and T. Majima, J. Am. Chem. Soc., 2011, 133, 7197–7204 CrossRef CAS PubMed.
- M. D'Arienzo, J. Carbajo, A. Bahamonde, M. Crippa, S. Polizzi, R. Scotti, L. Wahba and F. Morazzoni, J. Am. Chem. Soc., 2011, 133, 17652–17661 CrossRef PubMed.
- T. Ohno, K. Sarukawa and M. Matsumura, New J. Chem., 2002, 26, 1167–1170 RSC.
- X. Q. Gong and A. Selloni, J. Phys. Chem. B, 2005, 109, 19560–19562 CrossRef CAS PubMed.
- A. Selloni, Nat. Mater., 2008, 7, 613–615 CrossRef CAS PubMed.
- V. Shklover, M. K. Nazeeruddin, S. M. Zakeeruddin, C. Barbe, A. Kay, T. Haibach, W. Steurer, R. Hermann, H. U. Nissen and M. Gratzel, Chem. Mater., 1997, 9, 430–439 CrossRef.
- W. Hebenstreit, N. Ruzycki, G. S. Herman, Y. Gao and U. Diebold, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, R16334–R16336 CrossRef CAS.
- U. Diebold, N. Ruzycki, G. S. Herman and A. Selloni, Catal. Today, 2003, 85, 93–100 CrossRef CAS.
- G. S. Herman, Z. Dohnalek, N. Ruzycki and U. Diebold, J. Phys. Chem. B, 2003, 107, 2788–2795 CrossRef CAS.
- X.-Q. Gong, A. Selloni, M. Batzill and U. Diebold, Nat. Mater., 2006, 5, 665–670 CrossRef CAS PubMed.
- R. T. Zehr and M. A. Henderson, Surf. Sci., 2008, 602, 1507–1516 CrossRef CAS PubMed.
- Q. Wu, M. Liu, Z. Wu, Y. Li and L. Piao, J. Phys. Chem. C, 2012, 116, 26800–26804 CAS.
- W. Jiao, L. Wang, G. Liu, G. Q. Lu and H.-M. Cheng, ACS Catal., 2012, 2, 1854–1859 CrossRef CAS.
- O. Lamiel-Garcia, S. Tosoni and F. Illas, J. Phys. Chem. C, 2014, 118, 13667–13673 CAS.
- M. Setvin, X. Hao, B. Daniel, J. Pavelec, Z. Novotny, G. S. Parkinson, M. Schmid, G. Kresse, C. Franchini and U. Diebold, Angew. Chem., Int. Ed., 2014, 53, 4714–4716 CrossRef CAS PubMed.
- R. Hengerer, L. Kavan, P. Krtil and M. Gratzel, J. Electrochem. Soc., 2000, 147, 1467–1472 CrossRef CAS PubMed.
- A. Tilocca and A. Selloni, J. Phys. Chem. B, 2004, 108, 19314–19319 CrossRef CAS.
- A. Vittadini, A. Selloni, F. P. Rotzinger and M. Gratzel, Phys. Rev. Lett., 1998, 81, 2954–2957 CrossRef CAS.
- C. Xu, W. Yang, Q. Guo, D. Dai, M. Chen and X. Yang, J. Am. Chem. Soc., 2014, 136, 602–605 CrossRef CAS PubMed.
- L. Kavan, M. Gratzel, S. E. Gilbert, C. Klemenz and H. J. Scheel, J. Am. Chem. Soc., 1996, 118, 6716–6723 CrossRef CAS.
- R. Chong, J. Li, X. Zhou, Y. Ma, J. Yang, L. Huang, H. Han, F. Zhang and C. Li, Chem. Commun., 2014, 50, 165–167 RSC.
- Y.-L. Zhao, C.-H. Wang, Y. Zhai, R.-Q. Zhang and M. A. Van Hove, Phys. Chem. Chem. Phys., 2014, 16, 20806–20817 RSC.
- D. Zhang, G. Li, X. Yang and J. C. Yu, Chem. Commun., 2009, 4381–4383 RSC.
- Q. Wu, M. Liu, Z. Wu, Y. Li and L. Piao, J. Phys. Chem. B, 2012, 116, 26800–26804 CAS.
- Z. Zhao, Z. Sun, H. Zhao, M. Zheng, P. Du, J. Zhao and H. Fan, J. Mater. Chem., 2012, 22, 21965–21971 RSC.
- G. Liu, H. G. Yang, X. Wang, L. Cheng, H. Lu, L. Wang, G. Q. Lu and H.-M. Cheng, J. Phys. Chem. B, 2009, 113, 21784–21788 CAS.
- X. Han, Q. Kuang, M. Jin, Z. Xie and L. Zheng, J. Am. Chem. Soc., 2009, 131, 3152–3153 CrossRef CAS PubMed.
- C. Z. Wen, J. Z. Zhou, H. B. Jiang, Q. H. Hu, S. Z. Qiao and H. G. Yang, Chem. Commun., 2011, 47, 4400–4402 RSC.
- S. Xie, X. Han, Q. Kuang, J. Fu, L. Zhang, Z. Xie and L. Zheng, Chem. Commun., 2011, 47, 6722–6724 RSC.
- F. Amano, O.-O. Prieto-Mahaney, Y. Terada, T. Yasumoto, T. Shibayama and B. Ohtani, Chem. Mater., 2009, 21, 2601–2603 CrossRef CAS.
- M. Sayed, P. Fu, H. M. Khan and P. Zhang, Int. J. Photoenergy, 2014, 490264 Search PubMed.
- H. Wu, J. Ma, Y. Li, C. Zhang and H. He, Appl. Catal., B, 2014, 152, 82–87 CrossRef PubMed.
- X. Wu, Z. Chen, G. Q. Lu and L. Wang, Adv. Funct. Mater., 2011, 21, 4167–4172 CrossRef CAS PubMed.
- W. Yang, J. Li, Y. Wang, F. Zhu, W. Shi, F. Wan and D. Xu, Chem. Commun., 2011, 47, 1809–1811 RSC.
- W. Sun, T. Peng, Y. Liu, W. Yu, K. Zhang, H. F. Mehnane, C. Bu, S. Guo and X.-Z. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 9144–9149 CAS.
- W. Sun, K. Sun, T. Peng, S. You, H. Liu, L. Liang, S. Guo and X.-Z. Zhao, J. Power Sources, 2014, 262, 86–92 CrossRef CAS PubMed.
- L. Etgar, P. Gao, Z. Xue, Q. Peng, A. K. Chandiran, B. Liu, M. K. Nazeeruddin and M. Graetzel, J. Am. Chem. Soc., 2012, 134, 17396–17399 CrossRef CAS PubMed.
- Y. Yu, X. Wang, H. Sun and M. Ahmad, RSC Adv., 2012, 2, 7901–7905 RSC.
- S. Lunell, A. Stashans, L. Ojamae, H. Lindstrom and A. Hagfeldt, J. Am. Chem. Soc., 1997, 119, 7374–7380 CrossRef CAS.
- J. S. Chen, D. Luan, C. M. Li, F. Y. C. Boey, S. Qiao and X. W. Lou, Chem. Commun., 2010, 46, 8252–8254 RSC.
- X. H. Yang, Z. Li, C. Sun, H. G. Yang and C. Li, Chem. Mater., 2011, 23, 3486–3494 CrossRef CAS.
- W. Wang, C. Lu, Y. Ni and Z. Xu, CrystEngComm, 2013, 15, 2537–2543 RSC.
- Z. Zheng, B. Huang, X. Qin, X. Zhang, Y. Dai, M. Jiang, P. Wang and M.-H. Whangbo, Chem.–Eur. J., 2009, 15, 12576–12579 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Commun., 2011, 47, 4532–4534 RSC.
- H. Li, Y. Zeng, T. Huang, L. Piao, Z. Yan and M. Liu, Chem.–Eur. J., 2012, 18, 7525–7532 CrossRef CAS PubMed.
- M. Liu, L. Piao, W. Lu, S. Ju, L. Zhao, C. Zhou, H. Li and W. Wang, Nanoscale, 2010, 2, 1115–1117 RSC.
- T. Taguchi, Y. Saito, K. Sarukawa, T. Ohno and M. Matsumura, New J. Chem., 2003, 27, 1304–1306 RSC.
- Y. Wang, H. Zhang, Y. Han, P. Liu, X. Yao and H. Zhao, Chem. Commun., 2011, 47, 2829–2831 RSC.
- H. Xu, S. Ouyang, P. Li, T. Kako and J. Ye, ACS Appl. Mater. Interfaces, 2013, 5, 1348–1354 CAS.
- L. Ye, J. Mao, J. Liu, Z. Jiang, T. Peng and L. Zan, J. Mater. Chem. A, 2013, 1, 10532–10537 CAS.
- A. Tsujiko, T. Kisumi, Y. Magari, K. Murakoshi and Y. Nakato, J. Phys. Chem. B, 2000, 104, 4873–4879 CrossRef CAS.
- L. Wang, L. Zang, J. Zhao and C. Wang, Chem. Commun., 2012, 48, 11736–11738 RSC.
- Y. Yang, G. Wang, Q. Deng, S. Kang, D. H. L. Ng and H. Zhao, CrystEngComm, 2014, 16, 3091–3096 RSC.
- J. Li, K. Cao, Q. Li and D. Xu, CrystEngComm, 2012, 14, 83–85 RSC.
- J. Mao, L. Ye, K. Li, X. Zhang, J. Liu, T. Peng and L. Zan, Appl. Catal., B, 2014, 144, 855–862 CrossRef CAS PubMed.
- Y. Miao and J. Gao, J. Solid State Chem., 2012, 196, 372–378 CrossRef CAS PubMed.
- V. Thanh-Khue, N. Cuong Ky and Y. S. Kang, Chem.–Eur. J., 2013, 19, 9376–9380 CrossRef PubMed.
- A. S. Barnard and L. A. Curtiss, Nano Lett., 2005, 5, 1261–1266 CrossRef CAS.
- A. S. Barnard, P. Zapol and L. A. Curtiss, Surf. Sci., 2005, 582, 173–188 CrossRef CAS PubMed.
- L. Wu, B. X. Yang, X. H. Yang, Z. G. Chen, Z. Li, H. J. Zhao, X. Q. Gong and H. G. Yang, CrystEngComm, 2013, 15, 3252–3255 RSC.
- Z. Lai, F. Peng, Y. Wang, H. Wang, H. Yu, P. Liu and H. Zhao, J. Mater. Chem., 2012, 22, 23906–23912 RSC.
- N. Cuong Ky, H. G. Cha and Y. S. Kang, Cryst. Growth Des., 2011, 11, 3947–3953 Search PubMed.
- H. Jiang, J. Xing, C. Wang and H. Yang, Chin. J. Chem., 2013, 31, 1503–1507 CrossRef CAS PubMed.
- X. Ding, H. Ruan, C. Zheng, J. Yang and M. Wei, CrystEngComm, 2013, 15, 3040–3044 RSC.
- H. Xu, P. Reunchan, S. Ouyang, H. Tong, N. Umezawa, T. Kako and J. Ye, Chem. Mater., 2013, 25, 405–411 CrossRef CAS.
- Q. Wu, Z. Wu, Y. Li, H. Gao, L. Piao, T. Zhang and L. Du, Chin. J. Catal., 2012, 33, 1743–1753 CrossRef CAS.
- Z. Zheng, B. Huang, J. Lu, X. Qin, X. Zhang and Y. Dai, Chem.–Eur. J., 2011, 17, 15032–15038 CrossRef CAS PubMed.
- L. Ye, J. Liu, L. Tian, T. Peng and L. Zan, Appl. Catal., B, 2013, 134, 60–65 CrossRef PubMed.
- Z. Tan, K. Sato, S. Takami, C. Numako, M. Umetsu, K. Soga, M. Nakayama, R. Sasaki, T. Tanaka, C. Ogino, A. Kondo, K. Yamamoto, T. Hashishin and S. Ohara, RSC Adv., 2013, 3, 19268–19271 RSC.
- T. Tachikawa, N. Wang, S. Yamashita, S. C. Cui and T. Majima, Angew. Chem., Int. Ed., 2010, 49, 8593–8597 CrossRef CAS PubMed.
- P. M. Oliver, G. W. Watson, E. T. Kelsey and S. C. Parker, J. Mater. Chem., 1997, 7, 563–568 RSC.
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed.
- Y.-F. Li, Z.-P. Liu, L. Liu and W. Gao, J. Am. Chem. Soc., 2010, 132, 13008–13015 CrossRef CAS PubMed.
- J. S. Lee and J. Jang, J. Ind. Eng. Chem., 2014, 20, 363–371 CrossRef CAS PubMed.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- L. Etgar, W. Zhang, S. Gabriel, S. G. Hickey, M. K. Nazeeruddin, A. Eychmueller, B. Liu and M. Graetzel, Adv. Mater., 2012, 24, 2202–2206 CrossRef CAS PubMed.
- C. Wang, X. Zhang and Y. Liu, Nanoscale, 2014, 6, 5329–5337 RSC.
- X. Han, X. Wang, S. Xie, Q. Kuang, J. Ouyang, Z. Xie and L. Zheng, RSC Adv., 2012, 2, 3251–3253 RSC.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- D. Y. C. Leung, X. Fu, C. Wang, M. Ni, M. K. H. Leung, X. Wang and X. Fu, ChemSusChem, 2010, 3, 681–694 CrossRef CAS PubMed.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- E. Cui and G. Lu, J. Phys. Chem. B, 2013, 117, 26415–26425 CAS.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- Q. Kuang, X. Zheng and S. Yang, Chem.–Eur. J., 2014, 20, 2637–2645 CrossRef CAS PubMed.
- L. Zhang, W. Wang, S. Sun, D. Jiang and E. Gao, Appl. Catal., B, 2015, 162, 470–474 CrossRef CAS PubMed.
- K. Wenderich, A. Klaassen, I. Siretanu, F. Mugele and G. Mul, Angew. Chem., Int. Ed., 2014, 53, 12476–12479 CAS.
- Y. Luan, L. Jing, Y. Xie, X. Sun, Y. Feng and H. Fu, ACS Catal., 2013, 3, 1378–1385 CrossRef CAS.
- Q. Wang, C. Chen, D. Zhao, W. Ma and J. Zhao, Langmuir, 2008, 24, 7338–7345 CrossRef CAS PubMed.
- Z. He, Q. Cai, F. Hong, Z. Jiang, J. Chen and S. Song, Ind. Eng. Chem. Res., 2012, 51, 5662–5668 CrossRef CAS.
- J. Zhu, S. Wang, Z. Bian, S. Xie, C. Cai, J. Wang, H. Yang and H. Li, CrystEngComm, 2010, 12, 2219–2224 RSC.
- K. L. Ding, Z. J. Miao, Z. M. Liu, Z. F. Zhang, B. X. Han, G. M. An, S. D. Miao and Y. Xie, J. Am. Chem. Soc., 2007, 129, 6362–6363 CrossRef CAS PubMed.
- D. Zhang, G. Li, H. Wang, K. M. Chan and J. C. Yu, Cryst. Growth Des., 2010, 10, 1130–1137 CAS.
- W.-J. Lee and Y.-M. Sung, Cryst. Growth Des., 2012, 12, 5792–5795 CAS.
- L. Ye, J. Liu, Z. Jiang, T. Peng and L. Zan, Nanoscale, 2013, 5, 9391–9396 RSC.
- M. Kong, Y. Li, X. Chen, T. Tian, P. Fang, F. Zheng and X. Zhao, J. Am. Chem. Soc., 2011, 133, 16414–16417 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |