
Band engineering and charge separation in the Mo1−xWxS2/TiO2 heterostructure by alloying: first principle prediction†
Abstract
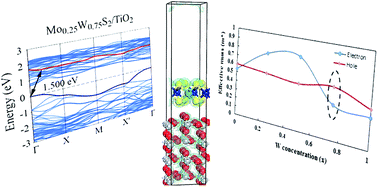
The composition dependent electronic properties of the Mo1−xWxS2 monolayer deposited over a TiO2 (110) substrate were investigated based on ab initio density functional calculations by applying periodic boundary conditions. Adsorption of the Mo1−xWxS2 monolayer on the TiO2 is physical in nature based on calculated binding energy values. It was found that the mechanical strain generated by the presence of TiO2 beneath the Mo1−xWxS2 layer resulted in a shift in band position of the Mo1−xWxS2 in favor of photoelectrochemical water splitting. Moreover, variation of W concentration in Mo1−xWxS2 could improve the charge separation and increase the effective mass ratio leading to an extension of the electron–hole pair lifetime which is useful for photocatalytic and photoelectrochemical applications.
 Please wait while we load your content...
Please wait while we load your content...

