
Mixed-solvothermal synthesis, structures, surface photovoltage, luminescence and molecular recognition properties of three new transition metal phosphonates with 3D framework and supramolecular structures†
Abstract
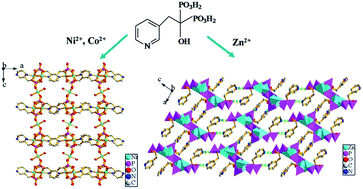
Three new transition metal(II) phosphonates with 3D framework and supramolecular structures, namely, [M1.5(L)(H2O)]·NH2(CH3)2·H2O (M = Ni (1), Co (2)) and [Zn2(H2L)(HL)]·NH2(CH3)2·3H2O (3) (H4L = C5H4NCH2C(OH)(PO3H2)2), have been synthesized under mixed-solvothermal conditions and structurally characterized. Compounds 1 and 2 are isostructural and adopt a 3D framework structure. The {M(1)O5N} octahedra and {CPO3} tetrahedra are interconnected into a 1D chain via corner-sharing, which is further linked to adjacent chains through pyridyl rings to form a 2D layer structure. Neighboring layers are bridged through {M(2)O6}, leading to a 3D framework structure with a 1D channel system along the b-axis. For compound 3, the {Zn(1)O4} and {Zn(2)O4} polyhedra are interconnected by phosphonate groups into a 1D double chain, and the double chain is further connected to adjacent chains through hydrogen bonds to form a 2D supramolecular network. Then these neighboring layers are further connected through hydrogen bonding interactions to give rise to a 3D supramolecular structure. Surface photovoltage spectroscopy (SPS) and field-induced surface photovoltage spectroscopy (FISPS) of compounds 1 and 2 indicate that they possess a positive SPV response in the range of 300–800 nm and show p-type semiconductor characteristics. Compound 3 has been realized for the sensitive sensing of N,N-dimethylformamide (DMF) by a luminescent method.
 Please wait while we load your content...
Please wait while we load your content...

