
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel hybrid Sr-doped TiO2/magnetic Ni0.6Zn0.4Fe2O4 for enhanced separation and photodegradation of organics under visible light
Fuming
Liu
a,
Yu
Xie
*a,
Changlin
Yu
c,
Xiaoming
Liu
a,
Yuhua
Dai
a,
Lianjun
Liu
*b and
Yun
Ling
a
aDepartment of Materials Chemistry, Nanchang Hangkong University, Nanchang, Jiangxi Province 330063, PR China. E-mail: xieyu_121@163.com; Fax: +86 791 83953373; Tel: +86 791 83953373
bMechanical Engineering Department, University of Wisconsin–Milwaukee, Milwaukee, WI 53211, USA. E-mail: liul@uwm.edu; Tel: +1 216 650 9842
cSchool of Metallurgy and Chemical Engineering, Jiangxi University of Science and Technology, Ganzhou, Jiangxi Province 341000, PR China
First published on 18th February 2015
Abstract
Titanium dioxide (TiO2) has been intensively used as a photocatalyst for the degradation of organic pollutants in water, but is typically obstacle by a low efficiency, costly separation, limited visible light response, and poor recyclability. Herein, we provided a reliable method to simultaneously tackle these four obstacles by developing an integrated and multifunctional hybrid photocatalyst/magnetic material, i.e., Sr–TiO2/Ni0.6Zn0.4Fe2O4. This novel hybrid not only demonstrated a high efficiency (90–100%) and a good cycling performance (90% maintenance) for photodegradation of bisphenol A (BPA) under both UV and visible light irradiation, but it can also efficiently work at a wide pH range (4–10) and can be easily separated from water for reuse only by introducing an external magnetic field. The materials structure-to-activity correlation has also been investigated. It was found that doping Sr2+ and a coupling magnetic material with TiO2 could extend the visible light response and create active defects in TiO2, which were responsible for the nearly three times higher activity than that of commercial TiO2(P25) under visible light. On the other hand, doping excessive Sr2+ lowered the surface area, enlarged the crystalline size and caused particle aggregation; thus, leading to a decrease in photocatalytic activity of the hybrid. These further modifications in the hybrid materials can provide a competitive alternative to control the organic pollutants in waste water.
1. Introduction
Photodegradation of organic pollutants using photocatalysts (e.g., ZnO, TiO2, WO3, CdS) holds a great promise to purify water and has been investigated intensively over the last decade.1–3 Among the photocatalysts, TiO2 has received great interest because of its chemical stability, high photocorrosion resistance, non-toxicity, and low-cost.3–8 However, bare TiO2 has a very low quantum efficiency primarily because of fast recombination rates of electron and hole pair (e−–h+) and limited visible light responses (due to its wide band gap in the range of 3.0–3.2 eV).1,8 The low efficiency of TiO2 is a major barrier for commercializing this photocatalytic technology. Complicated separation of photocatalyst for regeneration and reuse may also impede its practical application, but this challenge is seldom addressed in the literature.9–11 Generally, a TiO2-based power was suspended in a solution for the photodegradation of organic pollutants, and it often required a filtration or centrifugation process to separate the photocatalyst from water. This process may increase the cost and brings a potential genotoxicity due to some residual organics adsorbed on the solids.2,12 Therefore, it is urgent and necessary to develop high performance and easily recycled TiO2-based photocatalysts.To improve the photocatalytic efficiency of TiO2, great efforts have been made to modify TiO2 either by incorporating noble metals (e.g., Ag) or metal oxides (e.g., Fe2O3) to act as electron traps and inhibit the recombination of e−–h+ pairs,2,8,13–16 or by doping nonmetals (e.g., N) or co-doping with nonmetals and transition metal ions (e.g., Cr3+, Cu2+, Co2+, Mg2+) to extend the visible light response.9,17–23 Alternatively, pure or modified perovskite strontium titanate (SrTiO3) and SrTiO3/TiO2 heterojunctions have also been investigated as a candidate for the photodegradation of organic pollutants.23–27 However, a few studies have been conducted on Sr2+ doped TiO2 for the photodegradation of organic compounds. Very recently, Li et al.28 synthesized Sr2+-doped ZnO composites and attributed its enhanced performance over ZnO for RhB degradation under visible light to the narrowed band gap and the formation of active defects to trap electrons. Along this line, it is expected that doping Sr2+ into the TiO2 lattice will lead to an enhanced photocatalytic degradation of bisphenol A (BPA).
To overcome the obstacle of TiO2 powder separation from solution, several efforts have been attempted to anchor TiO2 on solid substrates (e.g., Ti mesh)4,11,29–33 or incorporate TiO2 with magnetic materials (e.g., ferrite).34–40 Immobilizing TiO2 on solid substrates is restricted to laboratory or small-scale applications because of the complicated preparation process, weak attachment of TiO2 on the foreign substrates, and deficient immersion and dispersion of TiO2 in the slurry solution. On the other hand, magnetic separation is a promising route to recover the used photocatalysts only by applying an external magnetic field. A number of studies have been attempted to fabricate core–shell structured magnetic material/photocatalysts for the photodegradation of organics, including Fe/TiO2/Ag, Fe/V/TiO2, nickel ferrite/N–TiO2, and strontium ferrite/N–TiO2.9,34,37,40 Unfortunately, the nanosized core magnetic materials are easily oxidized (e.g., Fe3O4 oxidized to Fe2O3) or rapidly transform in the crystal phase (e.g., γ-Fe2O3 ferromagnetic to α-Fe2O3 paramagnetic) when the calcination temperature is over 400 °C.34,40 Obviously, it is still a challenge to produce the TiO2-coated magnetic nanoparticles with concurrent good stability, high visible light activity and good magnetic properties.
To simultaneously overcome the challenges in the catalytic efficiency and separation of TiO2-based photocatalysts, in this work, we designed a novel hybrid by integrating a photocatalyst with magnetic material, i.e., Sr–TiO2/Ni0.6Zn0.4Fe2O4. We hypothesize that (1) doping TiO2 with Sr2+ could create some active defects and promote visible light absorption, and thus enhancing the charge separation and extending visible light response, (2) introducing Zn into NiFe2O4 could prevent the phase transition and assure good magnetic properties,41,42 and using Ni0.6Zn0.4Fe2O4 as magnetic core not only harvests visible light but also facilitates photocatalyst separation, and (3) an interface may be formed between Sr2+-doped TiO2 and magnetic Ni0.6Zn0.4Fe2O4, and thus inducing a synergistic effect to remove organics with an exceptional performance. To the best of our knowledge, for the first time this multifunctional material for water treatment has been developed. All the materials used (Ti, Sr, Ni, Zn, and Fe) to build the nanostructure are inexpensive and earth-abundant. The sol–gel method employed to prepare the hybrid is also simple and easy to scale-up. Another feature in this work is to evaluate the cycling performance of the hybrid photocatalyst/magnetic material under both UV and visible light irradiation, and attempt to establish the structure-to-activity relationships.
2. Materials and methods
2.1 Synthesis of magnetic Ni0.6Zn0.4Fe2O4 nanoparticles
Ni0.6Zn0.4Fe2O4 nanoparticles were prepared by a self-propagating combustion method. In brief, 0.1 mol nickel nitrate (Ni(NO3)2·6H2O), 0.067 mol zinc nitrate (Zn(NO3)2·6H2O) and 0.333 mol iron nitrate (Fe(NO3)3·9H2O) were dispersed into 100 ml deionized water with vigorous stirring. Citric acid (0.5 mol) was then added into the solution, using ammonium hydroxide to adjust the pH around 10.0. Next, the solution was heated in a water bath at 70 °C by microwave irradiation until the sol was formed. The as-received sol was finally dried in an oven at 100 °C for 24 h.2.2 Synthesis of hybrid Sr–TiO2/magnetic material
The hybrid Sr–TiO2/magnetic materials were prepared by a sol–gel method. Typically, in a beaker A, 10 ml of tetrabutyl titanate (C16H36O4Ti), 1 g of Ni0.6Zn0.4Fe2O4 nanoparticles, and 40 ml of absolute ethyl alcohol were uniformly mixed. In a beaker B, a certain amount of strontium nitrate (Sr(NO3)2) and starch (the mass ratio of Sr(NO3)2 to starch is 0.02![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5, 0.05:1, 0.1:1.5, 0.2:2.5), 5 ml de-ionized water, and 15 ml acetic acid were mixed. The mixture was heated for 5 min by microwave irradiation to get a homogeneous solution. Next, the solution in the beaker B was added dropwise to beaker A with mechanical stirring for 2 h. The mixed solution was aged at 70 °C for 24 h. The obtained gel was then dried and calcined at 550 °C in air for 4 h. The final hybrid materials were denoted as SrxTi/M (M = Ni0.6Zn0.4Fe2O4), where x is the nominal weight percentage of Sr2+ (i.e., 0.1 wt%, 0.25 wt%, 0.5 wt%, and 1 wt%). For comparison, Ti/M and Sr0.25Ti were also prepared using a same procedure as SrTi/M.
:0.5, 0.05:1, 0.1:1.5, 0.2:2.5), 5 ml de-ionized water, and 15 ml acetic acid were mixed. The mixture was heated for 5 min by microwave irradiation to get a homogeneous solution. Next, the solution in the beaker B was added dropwise to beaker A with mechanical stirring for 2 h. The mixed solution was aged at 70 °C for 24 h. The obtained gel was then dried and calcined at 550 °C in air for 4 h. The final hybrid materials were denoted as SrxTi/M (M = Ni0.6Zn0.4Fe2O4), where x is the nominal weight percentage of Sr2+ (i.e., 0.1 wt%, 0.25 wt%, 0.5 wt%, and 1 wt%). For comparison, Ti/M and Sr0.25Ti were also prepared using a same procedure as SrTi/M.
2.3 Characterization
The crystal structure of Sr–TiO2/Ni0.6Zn0.4Fe2O4 materials were characterized by X-ray diffraction (XRD) using a Bruker D8 Advance X-ray diffractometer with monochromated high-intensity CuKα radiation (λ = 0.15418 Å) in the 2θ range of 10–80°. The morphology and particle size of the materials were identified by field emission scanning electron microscopy (FESEM). Transmission electron microscopy (TEM) (JEOL TEM-3010) was used to approach the lattice fringes operating at an accelerating voltage of 300 keV. The surface area and porosity of the as-synthesized materials were examined by N2 adsorption/desorption at 77 K using the Brunauer–Emmett–Teller (BET) method (Micromeritics, ASAP 2020). The UV-vis diffuse reflectance spectra were recorded using a UV-vis-NIR spectrometer (Cary 5000, Varian). The valence states of Ti and O were identified by X-ray photoelectron spectroscopy (XPS), using a PHI 5000 versaprobe system using monochromatic Al KR radiation (1486.6 eV). All binding energies were referenced to the C 1s peak at 284.6 eV. The magnetic properties of the photocatalyst were evaluated at room temperature using a vibrating sample magnetometer (VSM, 9600-1 LDJ, USA) at a maximum applied field of 10 kOe.2.4 Photocatalytic activity measurement
The photocatalytic degradation of BPA was carried out in a hollow cylindrical photoreactor. The light source is a low-pressure mercury lamp (Beijing Ceaulight Co., Model CEL-LUV254, 10 W) that emits principally near 254 nm, or a long-arc xenon lamp (Beijing Ceaulight Co., Model CEL-LAX500, 500 W) to simulate visible light (>400 nm). The photoreactor was cooled by circulating water through a quartz channel inside, and the temperature was maintained at around 25 ± 2 °C. Prior to illumination, a 300 ml suspension with a certain amount of photocatalyst (0.5 g l−1) was stirred for 30 min to ensure the homogenous dispersion and full contact of BPA (10 ppm) with catalysts. The pH of the BPA solution was controlled using a NaOH solution (5 M, 1 M, 0.1 M) and an HNO3 solution (2 M. 0.1 M) to prepare basic and acidic solutions, respectively. After that, the lamp was turned on to irradiate the solution. A sample of 0.5 ml of solution was withdrawn at every 30 min. Moreover, the pH of the BPA solution was recorded at different reaction times using a pH meter (Mettler Toledo). The BPA concentration in each batch was measured by high-performance liquid chromatography (HPLC) equipped with a C-18 column (LUNA 5u 100A, 4.6 mm × 250 mm, Phenomenex) and a diode array detector (SPD-M20A, Shimadzu). The isocratic methanol–water mixture (70:30, v/v) as an eluent was employed at a flow rate of 1 ml min−1. The residual BPA content in the aqueous solution was determined with a standard curve (R2 = 0.9999) using a standard BPA solution for the calibration (i.e., 0, 0.2, 0.5, 1, 2, 5, 10 and 15 ppm). The degradation efficiency (DE) of BPA was calculated by the following equation: DE = Ct/Co, where Co and Ct are the initial and residual BPA concentration (ppm) at different reaction times, respectively.
3. Results and discussion
3.1 Crystal structure, morphology and texture
The crystal structure of the hybrid was identified by XRD. As shown in Fig. 1, all the samples displayed the same diffraction peaks, which are indexed to a mixture of the anatase phase of TiO2 (JCPDS no. 21-1272) and the spinel phase of Ni0.6Zn0.4Fe2O4.43,44 No additional peaks for perovskite SrTiO3 or SrO crystallites appeared because of the low calcination temperature and low dopant concentration of Sr2+.23,27 Noticeably, Ti/M without Sr2+ dopant exhibited sharp diffraction peaks. By contrast, SrxTi/M showed broadened and weakened diffraction peaks for both the TiO2 (101) plane and magnetic Ni0.6Zn0.4Fe2O4. This comparison indicated that doping Sr2+ ions into the TiO2 lattice could stabilize the crystal phase of TiO2 anatase and inhibit the aggregation and growth of the particles, thus inducing a decrease in TiO2 crystalline size. Among the samples, Sr0.1Ti/M showed the smallest crystalline size (see Table 1). Increasing the concentration of Sr2+ ions resulted in an increase in the TiO2 crystalline size from 15.7 nm to 18.8 nm, probably because the excessive Sr2+ ions cannot be completely doped into the TiO2 lattice and caused particle agglomeration. Moreover, the uniformly dispersed Sr–TiO2 particles may fully attach on the surface of Ni0.6Zn0.4Fe2O4 and result in a decrease in its peak intensity. | ||
| Fig. 1 XRD patterns for Sr2+-doped TiO2/Ni0.6Zn0.4Fe2O4 nanocomposites with a series of concentrations of Sr2+. | ||
| Sample name | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) | Crystallite size of TiO2 (nm) |
|---|---|---|---|---|
| Sr0.1Ti/M | 64 | 0.205 | 12.7 | 15.7 |
| Sr0.25Ti/M | 66 | 0.198 | 12.0 | 15.9 |
| Sr0.5Ti/M | 44 | 0.063 | 5.6 | 17.5 |
| Sr1.0Ti/M | 46 | 0.071 | 6.1 | 18.8 |
The morphology and particle size of the SrxTi/M samples were characterized by SEM (Fig. 2) and TEM (Fig. 3). As shown in Fig. 2, Sr0.1Ti/M and Sr0.2Ti/M have uniformly dispersed particles with an average size of 20 nm. By contrast, Sr1.0Ti/M has randomly mixed small particles and big grains (over 100 nm). The TEM image at a low magnification in Fig. 3a further confirmed that Sr0.1Ti/M was composed of relatively uniform spherical or rectangle particles with a diameter of ∼20 nm. The TEM image at a high magnification in Fig. 3b clearly shows well-faceted TiO2 nanocrystals with an interplanar spacing of 0.352 nm that matches the (101) plane of anatase phase.5,45,46 Ni0.6Zn0.4Fe2O4 nanoparticles (∼15 nm), located beyond TiO2 particles, was also observed with a lattice spacing of 0.187 nm.47 Again, SEM and TEM images directly supported the XRD observations that a small amount of Sr2+ (below 0.25 wt%) doped TiO2 particles were well-patched onto the magnetic Ni0.6Zn0.4Fe2O4, while excessive Sr2+ dopant easily induced particles aggregation into large grains.
 | ||
| Fig. 2 SEM images for (a) Sr0.1Ti/M, (b) Sr0.25Ti/M, and (c) Sr1.0Ti/M composites (M = Ni0.6Zn0.4Fe2O4). | ||
 | ||
| Fig. 3 (a) TEM and (b) HRTEM images for Sr0.1Ti/M (M = Ni0.6Zn0.4Fe2O4). | ||
Table 1 also compared the measured BET surface area, pore size, pore volume of the SrxTi/M samples to explore the effect of Sr2+-dopant content on the textural property. The presence of mesopores in SrxTi/M could result from the space between particles, as evidenced by the SEM images in Fig. 2. At low loadings of Sr2+, Sr0.1Ti/M and Sr0.25Ti/M displayed similar surface areas, pore sizes, and pore volumes, which were considerably higher than those of Sr0.5Ti/M and Sr1.0Ti/M at high loadings of Sr2+. The data in Table 1 agrees well with XRD and SEM observations that high contents of Sr2+ results in the aggregation of the particles and the growth in crystalline size, which could block the micro/meso-pores within TiO2 particles and decrease the BET surface area, pore size, and pore volume.
3.2 Optical property and chemical state
UV-vis spectra, displayed in absorbance units, have been recorded to investigate the light response of the SrxTi/M samples. As shown in Fig. 4, the absorption edge of bare TiO2(P25) is located around 400 nm, corresponding to a band gap of about 3.10 eV. By contrast, incorporating the magnetic Ni0.6Zn0.4Fe2O4 with TiO2 induced the appearance of a tail extending to a longer wavelength of 600 nm. All the SrxTi/M samples display similar broad bands extending to nearly 800 nm, which is consistent with the dark gray physical appearance of the samples. Furthermore, the band edge of Sr0.1Ti/M red shifted to about 520 nm, corresponding to a small band gap of 2.4 eV. The UV-vis results confirmed our original hypothesis that doping Sr2+ into TiO2 and coupling it with magnetic Ni0.6Zn0.4Fe2O4 could narrow the band gap of TiO2 and harvest visible light.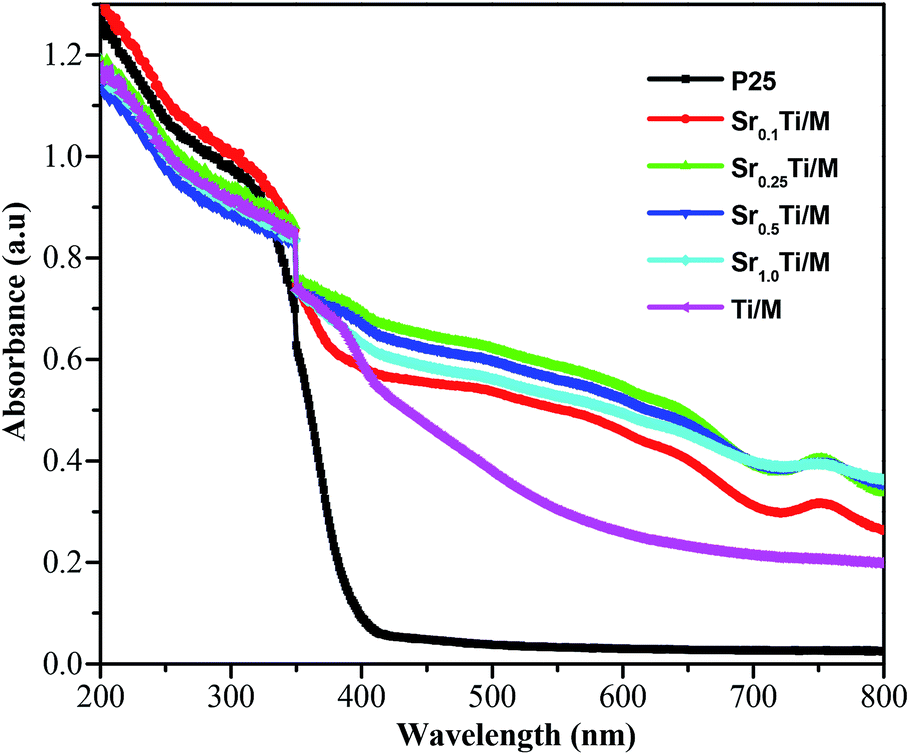 | ||
| Fig. 4 UV-vis spectra for Sr2+-doped TiO2/Ni0.6Zn0.4Fe2O4 nanocomposites with different Sr2+ concentrations. | ||
XPS has been conducted to approach the chemical states of TiO2 with and without doping Sr. As shown in Fig. 5a, Ti/M displayed Ti 2p3/2 and the Ti 2P1/2 binding energies at 458.6 and 464.2 eV, respectively, corresponding to a typical characteristic of the Ti4+ oxidation state.16,40 Interestingly, the Ti 2p binding energies of Sr0.1Ti/M slightly shifted to a lower level (by 0.4 eV). Similarly, doping Sr2+ into TiO2 also led to a chemical shift in the O 1s binding energy at 529.9 eV that is associated with lattice oxygen (Ol) bonded to metal ions, while the O 1s related to surface oxygenated species (Oa) (e.g., OH groups, adsorbed H2O) remained at a same position at 531.5 eV.45 Moreover, the relative amount of Ol (by calculating the peak area ratio of Ol/(Ol + Oa) in Fig. 5b) in Sr0.01Ti/M (78.6%) was slightly higher than that in Ti/M (74.8%). Generally, the chemical shifts in XPS spectra resulted from either the formation of a new oxidation state or the changes in the local chemical and physical environment. Because no shoulder peak for the new state of Ti3+ was observed in Sr0.1Ti/M, the slight chemical shift is primarily related to a change in the local chemical environment of TiO2. In other words, Sr2+ has been successfully doped into TiO2 lattice. Because some Ti4+ sites have been substituted by Sr2+ ions, some defects (e.g., oxygen vacancy, VO) were formed for charge compensation.16,29 This is also supported by the XPS quantitative analysis that Ti/M has a higher ratio of O/(Ti + Zn + Ni + Fe) (2.61) than does Sr0.1Ti/M (2.02). Hence, the change in the chemical environment of TiO2 was possibly caused by the partial replacement of Ti4+ by Sr2+ dopants and the formation of VO in TiO2.
 | ||
| Fig. 5 XPS spectra for undoped TiO2/Ni0.6Zn0.4Fe2O4 and Sr0.1Ti/Ni0.6Zn0.4Fe2O4 composites: (a) Ti 2p, and (b) O 1s. | ||
3.3 Photocatalytic performance measurement
Photocatalytic degradation of BPA over the hybrid SrxTi/M was first conducted under UV (254 nm) irradiation. Fig. 6 compares the photodegradation efficiency of various SrxTi/M. Right before the photodegradation, a background experiment was carried out without UV irradiation in the presence of Sr0.1Ti/M. The result showed that in the dark process, the concentration of BPA almost remained same as the initial one within 4 h, indicating that no photocatalytic reaction took place without photo-illumination. At low contents of Sr2+, Sr0.1Ti/M and Sr0.25Ti/M exhibited similar photodegradation activity. Furthermore, increasing the concentration of Sr2+ resulted in a decrease in the activity over Sr0.5Ti/M and Sr1.0Ti/M. By correlating the structure and property of the materials (see the results in Table 1, Fig. 1 and 2), the inferior activity at a high content of Sr2+ is probably because Sr0.5Ti/M and Sr1.0Ti/M have larger crystalline sizes, smaller specific surface areas and pore volumes, and less uniform particle dispersions. These drawbacks on Sr0.5Ti/M and Sr1.0Ti/M could hinder the adsorption and activation of BPA on the surface. Excessive Sr2+ may also induce the formation of a new e−–h+ recombination center that shortened the lifetime of photogenerated charge carriers.28 Another important result in Fig. 6 is that Sr0.1Ti/M and Sr0.25Ti/M displayed a considerably higher efficiency compared to Ti/M, Sr0.25Ti, commercial TiO2(P25), and bare M. Sr0.1Ti/M is able to eliminate nearly 100% BPA at 4 h, while TiO2(P25) and bare M can only reach 40% and 25%, respectively. In addition, Sr0.25Ti and Ti/M was even more active than bare TiO2(P25) and M. In this regard, it is believed that there is a synergy between Sr2+ dopant and magnetic M that remarkably prevents the charge recombination in TiO2, as a consequence of leading to a superior activity of Sr0.1Ti/M. | ||
| Fig. 6 Photodegradation of BPA by various Sr2+-doped TiO2/M (M = Ni0.6Zn0.4Fe2O4) catalysts under UV-254 nm irradiation as a function of time (catalyst dosage = 0.5 mg ml−1, initial BPA concentration = 10 ppm, near neutral pH). | ||
We also performed photocatalytic degradation of BPA at different pH conditions using a Sr0.25Ti/M composite under UV irradiation because the waste water may be acidic or basic. The initial pH of the reactant solution was controlled by adding a desired amount of NaOH or HNO3. Fig. 7a compares the photocatalytic performance of Sr0.25Ti/M in a pH range of 4–10. It was found that either at basic (pH = 10) or acidic conditions (pH = 4), Sr0.25Ti/M displayed a higher efficiency that at near neutral conditions (pH = 6–8) within the beginning 2 h, while the efficiency eventually remained almost the same at the later stage and reached as high as nearly 99% at 3.5 h in the entire pH range (4–10). This result strongly suggested that our novel hybrid SrxTi/M is able to efficiently work at a wide pH range. Moreover, the pH evolution during the photocatalytic reaction was also monitored, as shown in Fig. 7b. At a basic condition, the pH gradually decreased and reached a steady state to near neutral, while at an acidic condition, the initial pH gradually increased. If starting with a near neutral solution, the pH only slowly decreased. The evolution of solution pH revealed that both proton (H+) and OH˙ radicals play a critical role in the photodegradation of BPA. A possible mechanism has been proposed to explain the pH impacts, as shown in Fig. 8. The starting solution with a high pH contains more OH− anions, which could act as hole (h+) scavengers and react with photo-generated h+ to form active OH˙ radicals (Fig. 8a).16,27 Hence, the consumption of OH− led to the decrease in pH over the reaction time. The OH˙ radicals have a strong oxidation ability to degrade BPA to CO2 and H2O, which could explain why the activity is higher at pH = 10. On the other hand, the starting solution with a low pH contains more H+ ions, which serves as electron scavengers and inhibits the recombination of e−–h+ pairs (Fig. 8b). Moreover, the H˙ radicals, generated from H+ + e− → H˙, could react with O2 to form hydrogen peroxide that decomposes into oxidative OH˙ under UV irradiation.48 Thus, the suppressed recombination and the formation of OH˙ leads to an increase in pH over time and a higher efficiency than at neutral pH values.
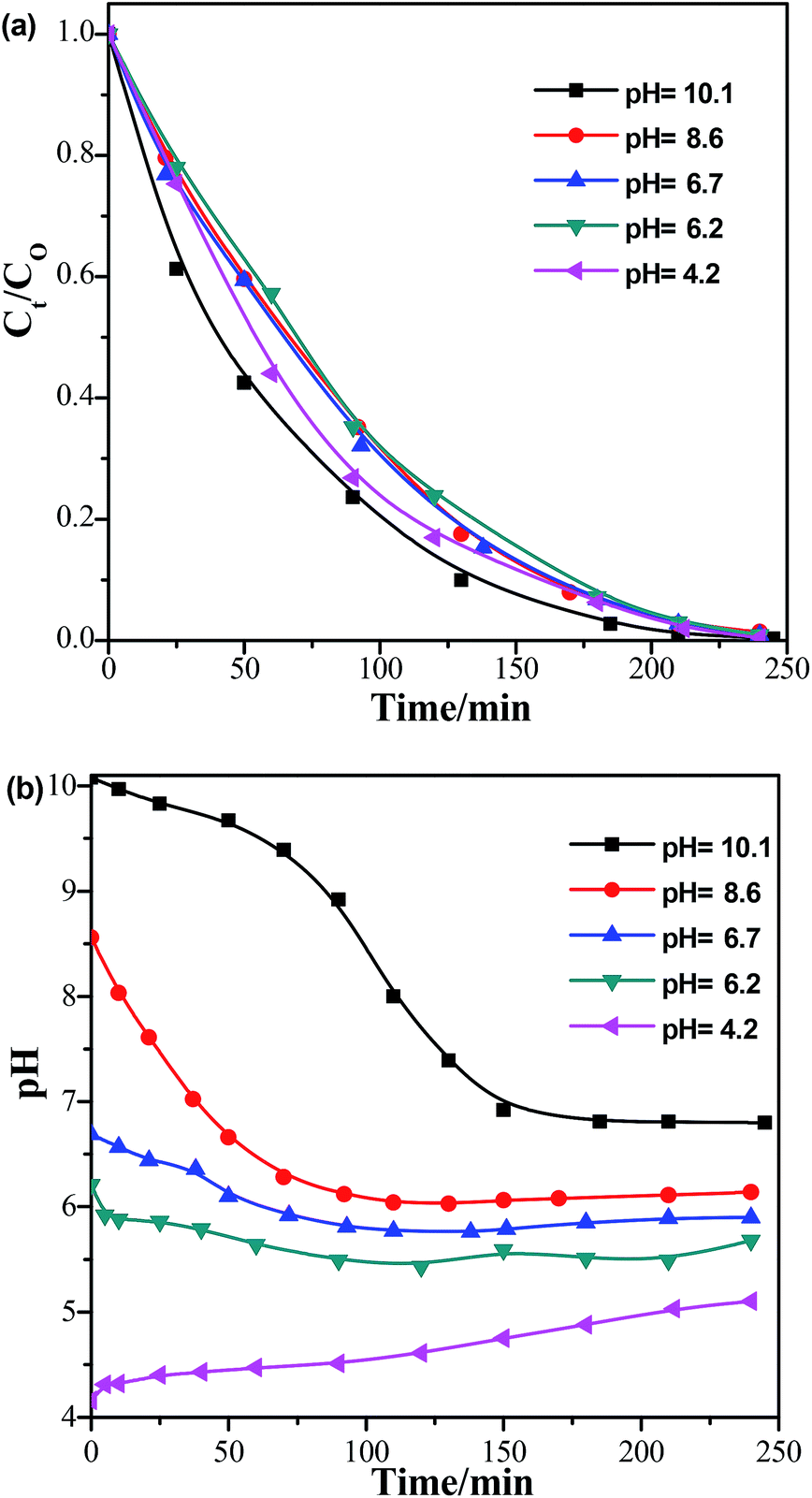 | ||
| Fig. 7 Photodegradation of BPA by Sr0.25Ti/M (M = Ni0.6Zn0.4Fe2O4) under UV-254 nm irradiation (a) at different pH environments and (b) the evolution of pH as a function of time (catalyst dosage = 0.5 mg ml−1, initial BPA concentration = 10 ppm). | ||
 | ||
| Fig. 8 The possible mechanism for BPA photodegradation under (a) basic and (b) acidic conditions. | ||
The photocatalytic performance of the SrxTi/M samples was also evaluated under visible light (400–1000 nm) irradiation, as shown in Fig. 9. The order of activity of the hybrids was Sr0.1Ti/M > Sr0.25Ti/M ≈ Sr0.5Ti/M ≈ Sr1.0Ti/M > Sr0.25Ti > Ti/M > P25 > M, which is very similar to the trend under UV irradiation (see Fig. 6). Even under visible light irradiation, the photodegradation efficiency of Sr0.1Ti/M could reach as high as 90% at 4 h, nearly 4 times higher than bare TiO2(P25) and M (only 15–20%). In addition to the larger surface area, smaller crystalline size, and more uniform particle dispersion, the superior performance of Sr0.1Ti/M under visible light is probably also due to another two reasons. First, incorporating an appropriate amount of Sr2+ dopant and magnetic M with TiO2 narrowed its band gap (see UV-vis spectra in Fig. 4); thus, enhancing the harvest and utilization of visible light. Second, as evidenced by XPS results, doping Sr2+ into TiO2 also created some active defect sites (oxygen vacancies), which may induce the formation of the new energy state located below the conduction band minimum of TiO2. The active defect sites could facilitate charge separation and trap electrons even under visible light.16,28,49 The electrons accumulated at defect sites could easily attach on O2 molecules to produce powerful superoxide radicals (˙O2−) that promote the activation and oxidation of BPA molecules.
 | ||
| Fig. 9 Photodegradation of BPA by various Sr2+-doped TiO2/M (M = Ni0.6Zn0.4Fe2O4) catalysts under visible light irradiation (400–1000 nm) as a function of time (catalyst dosage = 0.5 mg ml−1, initial BPA concentration = 10 ppm, near neutral pH). | ||
3.4 Material separation and cycling performance
The above photocatalytic activity results indicated that among all the tested samples, Sr0.1Ti/M has an outstanding performance under both UV and visible light irradiation. To further understand its superiority, we measured the cycling performance of Sr0.1Ti/M during three runs of photodegradation alternations. In between each cycle, the spent Sr0.1Ti/M was separated only by adding an external magnetic field around the solution and washed with water without any high temperature treatment or centrifugation. Above all, we measured the magnetic property of Sr0.1Ti/M at room temperature using vibrating magnetometer. Fig. 10 shows the magnetization (M)–magnetic field (H) loop of Sr0.1Ti/M. It was found that Sr0.1Ti/M exhibited a typical soft ferrite behavior with a saturated magnetization (Ms) of 19.04 emu g−1, the number of which was lower than the literature reported for pure Ni0.6Zn0.4Fe2O4.42,43 This is because our Ni0.6Zn0.4Fe2O4 was encapsulated with nonmagnetic Sr0.1TiO2 nanoparticles. Although Sr0.1Ti/M has a relatively low Ms, it still can be easily magnetically separated for reuse. As evidenced by the inset photograph in Fig. 10, the suspended solution quickly became clear once placing a magnet near the bottle wall for 30 s, and the powder was accumulated and attached on the wall. | ||
| Fig. 10 Room temperature hysteresis behaviour of Sr0.1Ti/M (M = Ni0.6Zn0.4Fe2O4). Inset photograph shows a separation process of Sr0.1Ti/M from aqueous solution using a magnet. | ||
Fig. 11 compares the photodegradation efficiency of Sr0.1Ti/M in each cycle under both UV and visible light irradiation after 4 h. After three cycles, the efficiency in the 3rd run only slightly dropped at both conditions, but still maintained as high as 89% and 78% under UV and visible light irradiation, respectively. Obviously, Sr0.1Ti/M demonstrated a good stability and recyclability, where M could provide a magnetic field for separation and harvest visible light concurrently. This good cycling performance also confirms our original hypothesis that integrating metal ion doped photocatalysts (e.g., Sr–TiO2) and magnetic materials (e.g., Ni0.6Zn0.4Fe2O4) is a reliable and convenient method to simultaneously enhance material separation and visible light activity for water purification.
 | ||
| Fig. 11 The cycling performance of Sr0.1Ti/M (M = Ni0.6Zn0.4Fe2O4) for photodegradation of BPA under UV-254 nm and visible light (>400 nm) irradiation (in each cycle, BPA concentration = 10 ppm, near neutral pH). | ||
4. Conclusions
In this work, we designed a novel hybrid photocatalyst/magnetic material, i.e., Sr2+ doped TiO2/Ni0.6Zn0.4Fe2O4, in order to enhance the efficiency of photo-degrading organic pollutants under visible light and to easily separate and reuse the material. The hybrid was synthesized by a simple sol–gel method. We found that doping a low concentration of Sr2+ (below 0.25 wt%) induced a smaller crystalline size, larger surface area and pore volume, and more uniform particle dispersion than did loading a high concentration of Sr2+. Moreover, doping Sr2+ ions and coupling a magnetic material with TiO2 narrowed the band gap and induced the generation of defect sites in TiO2. As a result, the integrated hybrid with a low loading of Sr2+ not only demonstrated a high efficiency (over 90%) and a good recycling performance (90% maintenance) under both UV and visible light irradiation, but also can efficiently work at a wide pH range (4–10) and be easily separated only by adding an external magnetic field. Furthermore, the hybrid Sr–TiO2/Ni0.6Zn0.4Fe2O4 showed an over two-times higher activity than those of TiO2/Ni0.6Zn0.4Fe2O4, commercial TiO2(P25) and bare Ni0.6Zn0.4Fe2O4, as well as 50% higher activity than that of Sr-doped TiO2, indicating that there is a synergy between the doped photocatalyst and magnetic material. The findings in this work suggest a new direction to engineer a smart photocatalyst/magnetic material heterojunction and control the interface between them, and it sheds light on the material's application in other aqueous-solid phase photocatalytic reactions.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (no. 51468043), Natural Science Foundation of Jiangxi Province (no. 20132BAB203018), Jiangxi Province Youth Scientists Cultivating Object Program (no. 20112BCB23017), Key Laboratory of Photochemical Conversion and Optoelectronic Materials, TIPC, CSA (no. PCOM201401).Notes and references
- S. Y. Lee and S. J. Park, J. Ind. Eng. Chem., 2013, 19, 1761–1769 CrossRef CAS PubMed.
- M. A. Patil and P. A. Parikh, Bull. Environ. Contam. Toxicol., 2014, 92, 109–114 CrossRef CAS PubMed.
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- C. Sriwong, S. Wongnawa and O. Patarapaiboolchai, J. Environ. Sci., 2012, 24, 464–472 CrossRef CAS.
- Z. Cheng, X. Quan, J. Xiang, Y. Huang and Y. Xu, J. Environ. Sci., 2012, 24, 1317–1326 CrossRef CAS.
- S. Lakshmi, R. Renganathan and S. Fujita, J. Photochem. Photobiol., A, 1995, 88, 163–167 CrossRef CAS.
- Y. Ohko, I. Ando, C. Niwa, T. Tatsuma, T. Yamamura, T. Nakashima, Y. Kubota and A. Fujishima, Environ. Sci. Technol., 2001, 35, 2365–2368 CrossRef CAS.
- E. Grabowska, J. Reszczynska and A. Zaleska, Water Res., 2012, 46, 5453–5471 CrossRef CAS PubMed.
- M. D'Arienzo, R. Scotti, L. Wahba, C. Battocchio, E. Bemporad, A. Nale and F. Morazzoni, Appl. Catal., B, 2009, 93, 149–155 CrossRef PubMed.
- P. Tian, H. Zhang, Z. Y. Shi, M. Y. Zhang, L. Wei and Z. Q. Yang, Applications of Engineering Materials, Pts 1–4, 2011, vol. 287–290, pp. 1815–1818 Search PubMed.
- V. M. Daskalaki, Z. Frontistis, D. Mantzavinos and A. Katsaounis, Catal. Today, 2011, 161, 110–114 CrossRef CAS PubMed.
- Z. Magdolenova, A. Collins, A. Kumar, A. Dhawan, V. Stone and M. Dusinska, Nanotoxicology, 2014, 8, 233–278 CrossRef CAS PubMed.
- S. C. Xu, Y. X. Zhang, S. S. Pan, H. L. Ding and G. H. Li, J. Hazard. Mater., 2011, 196, 29–35 CrossRef CAS PubMed.
- R. F. Yuan, B. H. Zhou, D. Hua and C. H. Shi, J. Hazard. Mater., 2013, 262, 527–538 CrossRef CAS PubMed.
- S. D. Kim, W. G. Choe and J. R. Jeong, Ultrason. Sonochem., 2013, 20, 1456–1462 CrossRef CAS PubMed.
- B. F. Gao, T. M. Lim, D. P. Subagio and T. T. Lim, Appl. Catal., A, 2010, 375, 107–115 CrossRef CAS PubMed.
- X. P. Wang, Y. X. Tang, M. Y. Leiw and T. T. Lim, Appl. Catal., A, 2011, 409, 257–266 CrossRef PubMed.
- R. H. Zhang, Q. Wang, J. Liang, Q. Li, J. F. Dai and W. X. Li, Phys. B, 2012, 407, 2709–2715 CrossRef CAS PubMed.
- D. Avisar, I. Horovitz, L. Lozzi, F. Ruggieri, M. Baker, M. L. Abel and H. Mamane, J. Hazard. Mater., 2013, 244, 463–471 CrossRef PubMed.
- C. H. Wu, C. Y. Kuo, C. J. Lin and P. K. Chiu, Int. J. Photoenergy, 2013, 2013, 1–9 Search PubMed.
- L. G. Devi and R. Kavitha, Appl. Catal., B, 2013, 140, 559–587 CrossRef PubMed.
- H. R. Rajabi, O. Khani, M. Shamsipur and V. Vatanpour, J. Hazard. Mater., 2013, 250, 370–378 CrossRef PubMed.
- I. R. Qazi, W.-J. Lee, H.-C. Lee, M. S. Hassan and O. B. Yang, J. Nanosci. Nanotechnol., 2010, 10, 3430–3434 CrossRef CAS PubMed.
- F. Zou, Z. Jiang, X. Qin, Y. Zhao, L. Jiang, J. Zhi, T. Xiao and P. P. Edwards, Chem. Commun., 2012, 48, 8514–8516 RSC.
- O. Ruzimuradov, S. Nurmanov, M. Hojamberdiev, R. M. Prasad, A. Gurlo, J. Broetz, K. Nakanishi and R. Riedel, Mater. Lett., 2014, 116, 353–355 CrossRef CAS PubMed.
- Y. Zhang, S. Lin, W. Zhang, H. Ge, G. Li, Y. Zhang, F.-Y. Qi and X.-M. Song, RCS Adv., 2014, 4, 3226–3232 CAS.
- L. Kumaresan, M. Mahalakshmi, M. Palanichamy and V. Murugesan, Ind. Eng. Chem. Res., 2010, 49, 1480–1485 CrossRef CAS.
- D. Li, J.-F. Huang, L.-Y. Cao, J.-Y. Li, H.-B. OuYang and C.-Y. Yao, Ceram. Int., 2014, 40, 2647–2653 CrossRef CAS PubMed.
- A. Zacharakis, E. Chatzisymeon, V. Binas, Z. Frontistis, D. Venieri and D. Mantzavinos, Int. J. Photoenergy, 2013, 2013, 1–9 CrossRef PubMed.
- N. Miranda-Garcia, S. Suarez, M. I. Maldonado, S. Malato and B. Sanchez, Catal. Today, 2014, 230, 27–34 CrossRef CAS PubMed.
- J. Hou, G. Dong, Y. Ye and V. Chen, J. Membr. Sci., 2014, 469, 19–30 CrossRef CAS PubMed.
- C.-Y. Kuo, C.-H. Wu and H.-Y. Lin, Environ. Technol., 2014, 35, 1851–1857 CrossRef CAS PubMed.
- E. M. Saggioro, A. S. Oliveira, T. Pavesi, M. Jimenez Tototzintle, M. Ignacio Maldonado, F. V. Correia and J. C. Moreira, Environ. Sci. Pollut. Res., 2014, 21, 12112–12121 CrossRef CAS PubMed.
- S. Xu, W. Shangguan, J. Yuan, M. Chen and J. Shi, Appl. Catal., B, 2007, 71, 177–184 CrossRef CAS PubMed.
- P. M. Álvarez, J. Jaramillo, F. López-Piñero and P. K. Plucinski, Appl. Catal., B, 2010, 100, 338–345 CrossRef PubMed.
- D. F. Sun, Y. D. Han, S. Gao and X. L. Zhang, Surf. Coat. Technol., 2013, 228, S516–S519 CrossRef CAS PubMed.
- M. Feyzi, H. R. Rafiee, S. Ranjbar, F. Jafari and B. Safari, Mater. Res. Bull., 2013, 48, 4844–4849 CrossRef CAS PubMed.
- Y. H. Tang, G. Zhang, C. B. Liu, S. L. Luo, X. L. Xu, L. Chen and B. G. Wang, J. Hazard. Mater., 2013, 252, 115–122 CrossRef PubMed.
- X. X. Yu, S. W. Liu and J. G. Yu, Appl. Catal., B, 2011, 104, 12–20 CrossRef CAS PubMed.
- A. Abd Aziz, K. S. Yong, S. Ibrahim and S. Pichiah, J. Hazard. Mater., 2012, 199, 143–150 CrossRef PubMed.
- K. Praveena, K. Sadhana, S. Srinath and S. R. Murthy, J. Phys. Chem. Solids, 2013, 74, 1329–1335 CrossRef CAS PubMed.
- Z. Wang, Y. Xie, P. Wang, Y. Ma, S. Jin and X. Liu, J. Magn. Magn. Mater., 2011, 323, 3121–3125 CrossRef CAS PubMed.
- V. D. Kapse, S. A. Ghosh, F. C. Raghuwanshi and S. D. Kapse, Mater. Chem. Phys., 2009, 113, 638–644 CrossRef CAS PubMed.
- T. Slatineanu, A. R. Iordan, M. N. Palamaru, O. F. Caltun, V. Gafton and L. Leontie, Mater. Res. Bull., 2011, 46, 1455–1460 CrossRef CAS PubMed.
- J. W. Ng, X. P. Wang and D. D. Sun, Appl. Catal., B, 2011, 110, 260–272 CrossRef CAS PubMed.
- L. Liu, D. T. Pitts, H. Zhao, C. Zhao and Y. Li, Appl. Catal., A, 2013, 467, 474–482 CrossRef CAS PubMed.
- B. Xu, B. Huang, H. Cheng, Z. Wang, X. Qin, X. Zhang and Y. Dai, Chem. Commun., 2012, 48, 6529–6531 RSC.
- C. G. Park, E. S. Choi, H. W. Jeon, J. H. Lee, B. W. Sung, Y. H. Cho and K. B. Ko, Desalin. Water Treat., 2014, 52, 797–804 CrossRef CAS.
- S. Ghasemi, S. Rahimnejad, S. R. Setayesh, S. Rohani and M. R. Gholami, J. Hazard. Mater., 2009, 172, 1573–1578 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |