
Determination of benzoylurea insecticides in environmental water and honey samples using ionic-liquid-mingled air-assisted liquid–liquid microextraction based on solidification of floating organic droplets†
Abstract
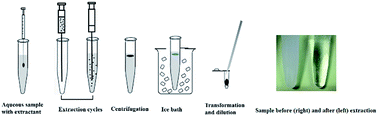
A novel and simple ionic-liquid-mingled air-assisted liquid–liquid microextraction based on solidification of floating organic droplets combined with high performance liquid chromatography was developed for the determination of six benzoylureas (BUs) in water and honey samples. In this method, a mixture of low-density and low melting point extraction solvents and aqueous sample solutions was rapidly sucked up and injected several times using a glass syringe. The influence of the main factors on the efficiency of this procedure is studied. Under the optimal conditions, the enrichment factors (EFs) for BUs were acquired in the range of 144 to 187, limits of detection (LODs) were between 0.01 and 0.1 μg L−1 and limits of quantitation (LOQs) were changed in the range of 0.03 and 0.33 μg L−1. The obtained extraction recoveries ranged from 84.03% to 109.20% with intra-day lower than 4.5%, and inter-day precision lower than 6.5%. The method is successfully applied to determine the BUs in environmental water and honey samples with recoveries in the range of 78.57–109.72%, which proved the potential use of this method in real samples.
 Please wait while we load your content...
Please wait while we load your content...

