DOI:
10.1039/C5RA00090D
(Paper)
RSC Adv., 2015,
5, 22510-22526
SAR studies of 3,14,19-derivatives of andrographolide on anti-proliferative activity to cancer cells and toxicity to zebrafish: an in vitro and in vivo study†
Received
3rd January 2015
, Accepted 10th February 2015
First published on 10th February 2015
Abstract
Andrographolide is bestowed with an interesting pharmacophore and has attracted numerous studies on the design and synthesis of andrographolide derivatives. In this study, a small library of 3,14,19-modified derivatives of andrographolide were synthesized and tested for their in vitro inhibitory activities to cancer cell growth and proliferation and in vivo toxicities against zebrafish embryo development. Structure anti-proliferative activity and toxicity relationships in current data revealed that the property of a substituent, substituted positions, and 14-stereochemistry together determined a compound's in vitro anti-proliferative activity against cancer cells, the in vivo toxicity to zebrafish, and the selectivity between MDA-MB-231 and A549. Taken together, our SAR studies discovered some potential leads for further development of anticancer drugs and suggested that the direct and/or indirect toxicity of an active compound with andrographolide pharmacophore should be given attention.
Introduction
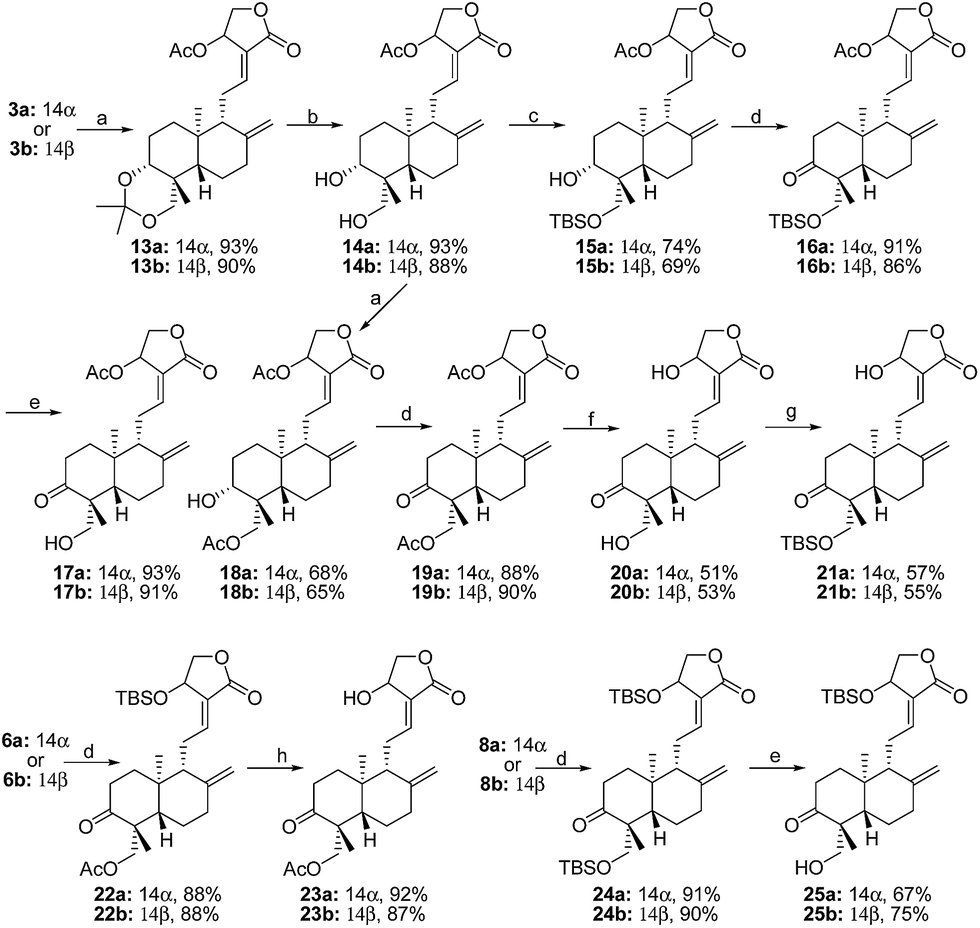
Andrographolide1 (1, Fig. 1), a labdane diterpene, is a representative ingredient of Andrographis paniculata (Burm.f.) Nees and plays an important role in “heat-clearing and detoxifying” defined in Chinese Medicine.2 Even though andrographolide obeys the “Rule of Five” and is bestowed with an interesting pharmacophore that displays various pharmacological activities and has therapeutic potential for a wide range of diseases,3 its poor water solubility and also relatively low lipo-solubility result in its weak potency and inadequate therapeutic efficacy and restrict its further application. To improve its physiochemical properties and pharmaceutical features, numerous andrographolide derivatives and their pharmacological activities have been reported in recent years, e.g., 14-acyloxy andrographolide derivatives and their antibacterial or/and anticancer activity,4 14-phenoxy andrographolide derivatives and their FXR antagonistic activity,5 dehydroandrographolide derivatives and their antiviral activity,6 and 19-tert-butyldimethylsilylated and 19-triphenylmethylated derivatives and their cytotoxic activities.4e Our interest was to discover 3,14,19-derivatives of andrographolide as potent anticancer cell proliferative agents and summarize SAR of a small library for the drug discovery and development of andrographolide.
 |
| | Fig. 1 Structures of andrographolide (1) and 14β-isomer (2) of andrographolide. | |
Zebrafish (Danio rerio) is an excellent in vivo model for physiologically relevant whole organism and behavior-based screening,7 which cannot be achieved with conventional in vitro systems. Along with the development of a zebrafish embryo, the activity and/or the toxicity of a tested compound reflects its degree of influence on the embryo development and integrity, and we have previously used a zebrafish model for the SAR analysis of seven polymethoxylated flavonoids for the anti-angiogenesis activity and toxicity.8 Moreover, our previous pilot study proved the concept that zebrafish is equipped with a sophisticated drug metabolism system and could address the challenge by predicting both potential toxicity and efficacy by considering the whole content of bioavailability, metabolism and multiple target effect of a hit.9 Because the pharmaceutical industry frequently encounters a high risk of failure in the development of a new drug candidate, particularly at later stages, owing to intolerable side-effects and/or toxicity in clinical trials, there is a trend in drug discovery strategy to exclude potentially toxic compounds at an early stage. On the basis of the feasibility of screening toxicity of compounds in a zebrafish model, our SAR study of andrographolide derivatives was performed by the discovery of their in vitro anticancer activities to cell proliferation of MDA-MB-231 and A549 and then the comparison of their in vivo toxicities against zebrafish embryo development.
In this paper, herein, a series of 3-, and/or 14-, and/or 19-modified derivatives of andrographolide as a small library were designed and synthesized. It was first discovered that some derivatives were in vitro active against the growth and proliferation of two cancer cell lines in an interesting structure–activity relationship. Further studies revealed the in vivo structure–toxicity relationship of these compounds against zebrafish embryo development. Interestingly, in contrast, either single or combined modifications at 3-, 14-, and 19-positions of the andrographolide scaffold caused a change in the anticancer proliferative activity and toxicity to zebrafish; moreover, the 14-stereochemistry also contributed to the anti-proliferative activity of cancer cells and toxicity to zebrafish to some extent, and the transformation between hydroxyl and ketone groups sometimes altered the anticancer activity and toxicity to zebrafish. Importantly, some specific modifications led to the selectivity between MDA-MB-231 and A549.
Results and discussion
The design of modifications and the preliminary results
In our initial study, it was discovered that andrographolide (1) (Table 1, entry 1) exhibited mild anti-proliferative effects on MDA-MB-231 and A549 and was not toxic to zebrafish; however, one derivative, 4a, of andrographolide (Table 1, entry 5) was not active against cancer cell proliferation of MDA-MB231 and A549 but showed a toxicity at a concentration of 300 μM against zebrafish embryo development. This observation drew our interest in 3,14,19-modifications of 1; thus, a series of andrographolide derivatives were designed, synthesized and tested for their in vitro anti-proliferative activities against cancer cells and their in vivo toxicities to zebrafish. In the design of these andrographolide derivatives, the intact core scaffold of 110 was used to explore whether and how the mono- or multi-modifications of 1 affected the anti-proliferative effect on cancer cells and toxicity to zebrafish.
Table 1 In vitro anti-proliferative activity to cancer cells after treatment with different compounds for 24 h and in vivo toxicity to zebrafish at 24 hpf
| Entry |
Cmpd |
C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logPa logPa |
CC50 (μM) |
Observation at concentrationb,c,d (μM) |
| MDA-MB-231 |
A549 |
10 |
30 |
100 |
300 |
| Calculated from Chem3D. “○” indicates that no toxicity was observed after zebrafish embryos were treated with the indicated concentration of the compound. “T” means that the indicated concentration of the compound treated with zebrafish embryos showed toxicity and the toxic effect was not determined. “D” indicates that zebrafish embryos treated with the indicated concentration of compound were dead. |
| 1 |
1 |
2.1186 |
109.7 ± 3.1 |
132 |
○ |
○ |
○ |
○ |
| 2 |
2 |
2.1186 |
98.1 ± 3.0 |
113.6 ± 18.1 |
○ |
○ |
○ |
○ |
| 3 |
3a |
4.1596 |
7.6 ± 0.3 |
37.2 ± 6.5 |
○ |
○ |
○ |
T |
| 4 |
3b |
4.1596 |
18.5 ± 3.1 |
71.7 ± 6.1 |
○ |
○ |
○ |
T |
| 5 |
4a |
7.5782 |
>300 |
>300 |
○ |
○ |
○ |
T |
| 6 |
4b |
7.5782 |
>300 |
>300 |
○ |
○ |
○ |
○ |
| 7 |
5a |
5.5372 |
4.8 ± 0.5 |
17.8 ± 3.2 |
○ |
○ |
○ |
○ |
| 8 |
5b |
5.5372 |
7.0 ± 0.3 |
10.5 ± 0.3 |
○ |
T |
D |
D |
| 9 |
6a |
6.4452 |
>300 |
>300 |
○ |
T |
T |
T |
| 10 |
6b |
6.4452 |
6.7 ± 0.2 |
56.5 ± 0.4 |
○ |
○ |
○ |
T |
| 11 |
7a |
7.3532 |
>300 |
>300 |
○ |
○ |
○ |
○ |
| 12 |
7b |
7.3532 |
133.2 ± 11 |
>300 |
○ |
○ |
○ |
T |
| 13 |
8a |
8.9142 |
>300 |
>300 |
○ |
○ |
○ |
○ |
| 14 |
8b |
8.9142 |
40.9 ± 4.8 |
120.2 ± 5.5 |
○ |
○ |
○ |
○ |
| 15 |
9a |
9.8222 |
>300 |
>300 |
○ |
○ |
○ |
○ |
| 16 |
9b |
9.8222 |
>300 |
>300 |
○ |
○ |
○ |
○ |
| 17 |
10a |
12.2912 |
>300 |
>300 |
○ |
○ |
○ |
○ |
| 18 |
10b |
12.2912 |
>300 |
>300 |
○ |
○ |
○ |
○ |
| 19 |
11a |
6.4452 |
4.7 ± 0.2 |
9.8 ± 0.3 |
○ |
T |
D |
D |
| 20 |
11b |
6.4452 |
4.3 ± 0.2 |
11.3 ± 0.4 |
○ |
T |
T |
D |
| 21 |
12a |
8.9142 |
>300 |
>300 |
○ |
○ |
○ |
○ |
| 22 |
12b |
8.9142 |
>300 |
>300 |
○ |
○ |
○ |
○ |
| 23 |
13a |
5.0182 |
7.6 ± 0.3 |
9.8 ± 0.3 |
○ |
T |
T |
T |
| 24 |
13b |
5.0182 |
4.0 ± 0.2 |
6.1 ± 0.2 |
○ |
D |
D |
D |
| 25 |
14a |
2.9772 |
10.2 ± 0.3 |
14.4 ± 0.2 |
○ |
○ |
○ |
T |
| 26 |
14b |
2.9772 |
8.7 ± 1.4 |
11.4 ± 2.1 |
○ |
○ |
○ |
○ |
| 27 |
15a |
6.3542 |
2.7 ± 0.1 |
3.3 ± 0.2 |
○ |
T |
T |
T |
| 28 |
15b |
6.3542 |
2.4 ± 0.2 |
2.5 ± 0.2 |
T |
D |
D |
D |
| 29 |
16a |
6.1014 |
2.4 ± 0.1 |
3.0 ± 0.2 |
○ |
T |
T |
T |
| 30 |
16b |
6.1014 |
2.7 ± 0.1 |
4.5 ± 0.2 |
T |
D |
D |
D |
| 31 |
17a |
2.7196 |
10.2 ± 0.1 |
5.6 ± 1.9 |
○ |
○ |
○ |
○ |
| 32 |
17b |
2.7196 |
6.1 ± 0.6 |
12.0 ± 2.0 |
○ |
○ |
○ |
○ |
| 33 |
18a |
3.8852 |
9.2 ± 1.4 |
14.4 ± 2.5 |
○ |
○ |
○ |
○ |
| 34 |
18b |
3.8852 |
3.9 ± 0.6 |
6.1 ± 1.4 |
○ |
○ |
○ |
○ |
| 35 |
19a |
3.6219 |
9.3 ± 0.3 |
10.8 ± 0.3 |
○ |
○ |
T |
T |
| 36 |
19b |
3.6219 |
3.7 ± 0.8 |
8.7 ± 1.7 |
○ |
T |
D |
D |
| 37 |
20a |
1.861 |
>300 |
>300 |
○ |
○ |
○ |
○ |
| 38 |
20b |
1.861 |
226.2 ± 4.7 |
>300 |
○ |
○ |
○ |
○ |
| 39 |
21a |
5.2428 |
>300 |
>300 |
○ |
○ |
○ |
○ |
| 40 |
21b |
5.2428 |
2.5 ± 0.5 |
5.9 ± 0.9 |
○ |
D |
D |
D |
| 41 |
22a |
6.1819 |
3.5 ± 0.2 |
8.9 ± 0.3 |
○ |
○ |
○ |
T |
| 42 |
22b |
6.1819 |
3.6 ± 0.6 |
21.6 ± 6.4 |
T |
D |
D |
D |
| 43 |
23a |
2.7633 |
27.2 ± 4.3 |
62.7 ± 9.4 |
○ |
○ |
○ |
○ |
| 44 |
23b |
2.7633 |
101.3 ± 8.4 |
168.5 ± 23.3 |
○ |
○ |
○ |
○ |
| 45 |
24a |
8.6614 |
>300 |
>300 |
○ |
○ |
○ |
○ |
| 46 |
24b |
8.6614 |
165.4 ± 6.0 |
>300 |
○ |
○ |
○ |
○ |
| 47 |
25a |
5.2796 |
10.8 ± 0.3 |
17.0 ± 0.1 |
○ |
○ |
T |
T |
| 48 |
25b |
5.2796 |
4.0 ± 0.5 |
10.7 ± 1.5 |
○ |
T |
D |
D |
3,14,19-Acylation and silylation are the most common reported modifications on andrographolide; thus, acetylation and silylation were chosen in this study as representatives to test in vitro anticancer cell proliferative activity and the in vivo zebrafish toxicity. The introduction of TBS (Scheme 1) or Ac (Scheme 2) generally increases a compound's lipophilicity, and these modifications can provide the correlation between the lipophilicity and the anticancer cell proliferative effect and/or zebrafish toxicity. In addition, 3-ketone derivatives (Scheme 2) were synthesized and tested because 3-ketone modification increases the lipophilicity and could change the possible binding mode due to the double bond. To know whether the 14-stereochemistry plays a role in exhibiting the anti-proliferative activity of cancer cells and/or causing toxicity to zebrafish, both the 14α- and 14β-isomers were synthesized and assayed.
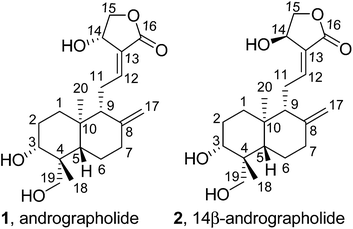
 |
| | Scheme 1 a Reagents and conditions: (a) TBSOTf, 2,6-lutidine, 0 °C, 0.5 h. (b) p-TSA, MeOH, 0 °C, 0.5 h. (c) AcCl, TEA, rt, 2 h. (d) Ac2O, cat. ZnCl2, 50 °C, 1 h. (e) TBSCl, imidazole, rt, 1 h. (f) for 9a and 9b: Ac2O, cat. DMAP, rt, 24 h. (g) for 10a and 10b: TBSOTf, 2,6-lutidine, 0 °C, 1 h. (h) from 9a and 9b to 11a and 11b: TFA/H2O (v/v = 10/1), −20 °C, 0.5 h. (i) from 10a and 10b to 12a and 12b: TFA, −20 °C, 5 min. | |
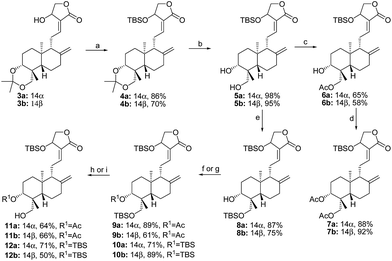
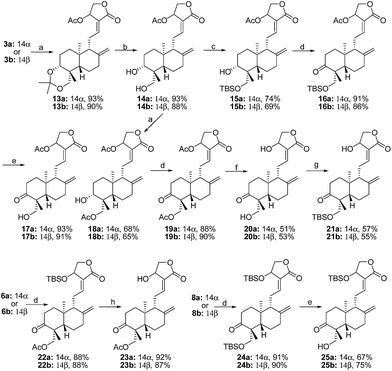 |
| | Scheme 2 a Reagents and conditions: (a) AcCl, TEA, rt, 1 h. (b) p-TSA, MeOH, 0 °C, 1 h. (c) TBSCl, TEA, rt, 1 h. (d) DMP, rt, 1 h. (e) TFA, −20 °C, 0.5 h. (f) p-TSA, MeOH, 40 °C, 8 h. (g) TBSCl, imidazole, rt, 1 h. (h) TBAF, THF, 0 °C, 3 h. | |
Synthesis
The transformation procedures of 3,14,19-O-substituted groups are depicted in Scheme 1. The preparation of 3a and stereochemistry transformation of 1 to 2 and 3b were achieved according to our previous report.5 14-O-Silylation of 3,19-acetonylidene-protected compounds 3a and 3b to obtain 4a11 and 4b, respectively, was accomplished at 0 °C using TBSOTf and 2,6-lutidine. The removal of 3,19-acetonylidene by p-TSA in methanol at 0 °C yielded 5a12 and 5b, which were selectively 19-O-acetylated using acetic chloride with TEA at room temperature to afford 6a and 6b.
The reaction of 6a and 6b with acetic anhydride catalyzed by anhydrous zinc chloride generated 3,19-diacetylated compounds 7a and 7b. Compounds 5a and 5b were converted to 14,19-di-TBS compounds 8a and 8b by TBSCl and imidazole. The reaction of 8a and 8b with acetic anhydride and DMAP (cat.) or with TBSOTf and 2,6-lutidine produced 9a and 9b or 10a and 10b, respectively, which were selectively hydrolyzed by 19-TBS group to afford 11a and 11b or 12a and 12b, respectively.
The synthetic routes of 3-ketones with 14,19-O-substituted groups are shown in Scheme 2. 14-Acetylation of 3a and 3b yielded 13a13 and 13b,5 followed by the selective hydrolysis of 3,19-acetonylidene to provide 14a13 and 14b.5 19-O-Silylation of 14a and 14b produced 15a14 and 15b, which were oxidized into 3-ketones 16a and 16b by Dess–Martin oxidation, and then the removal of 19-TBS protection of 16a and 16b by TFA at −20 °C gave 3-ketones 17a and 17b. 19-O-Acetylation of 14a and 14b produced 14,19-diacetylated 3-alcohol compounds 18a15 and 18b, and then the oxidation of 18a and 18b by DMP afforded 3-ketones 19a and 19b. After 19a and 19b were fully hydrolyzed into 20a16 and 20b by p-TSA in methanol at 40 °C, 20a and 20b were selectively 19-O-silylated to afford 21a and 21b. Compounds 23a and 23b were obtained through oxidation by DMP of 6a and 6b into 22a and 22b followed by 22a and 22b's deprotection of 14-OTBS using TBAF. Similarly, 10a and 10b were oxidized into 24a and 24b, and then the selective deprotection of 14-OTBS of 24a and 24b at −20 °C by TFA produced 25a and 25b.
Structure–anti-proliferative activity relationship on cancer cells and structure–toxicity relationship on zebrafish
Results of the in vitro anti-proliferative activity of cancer cells, the in vivo toxicity to zebrafish and the calculated ClogP values of the synthetic compounds are listed in Table 1. Andrographolide (1) and its 14β-epimer (2), which conform to the “Rule of Five” with a molecular weight of 350 Da, 3 hydrogen bond donors, 5 hydrogen bond acceptors and 2.1186 of the calculated ClogP, exhibited only weak anti-proliferation to two cancer cell lines (Table 1, entries 1 and 2) and showed no toxicity to zebrafish up to 300 μM (Table 1, entries 1 and 2). 3,19-Acetonylidene-protected compounds 3a and 3b (Table 1, entries 3 and 4) became more active against two cancer cell lines than their mother compounds 1 and 2, and they became toxic to zebrafish at the concentration of 300 μM, which could be derived partly from the increased hydrophobicity. Interestingly, 3a expressed two times more activity than 3b against the cell proliferation of two cancer cell lines. After the introduction of TBS at 14-position, both the compound 4a (Table 1, entry 5) and its 14β-epimer 4b (Table 1, entry 6) had no anti-cancer cell proliferative effect on MDA-MB-231 and A549, but 4a was toxic to zebrafish at 300 μM and 4b was non-toxic to zebrafish embryo development up to 300 μM. Unlike their mother compounds 4a and 4b, more hydrophilic 3,19-diol compounds 5a and 5b (Table 1, entries 7 and 8), which possess the marginal values of the “Rule of Five”, exhibited their anticancer cell proliferative activities with CC50 values of 4.8 and 17.8 μM, and 7.0 and 10.5 μM to MDA-MB-231 and A549, respectively. Moreover, 5a and 5b showed totally different toxicities to zebrafish as compared to 4a and 4b; the 14α-isomer 5a (Table 1, entry 7) was a safe compound for zebrafish up to 300 μM, but the 14β-isomer 5b (Table 1, entry 8) showed obvious toxicity to zebrafish at 30 μM and killed zebrafish at 100 μM, suggesting that the stereochemistry of 14-position herein played an important role in toxicity to zebrafish. The observation that anti-proliferative activity of cancer cells was increased from 1 and 2 to less hydrophilic 3a and 3b, and from 4a and 4b to more hydrophilic 5a and 5b, but decreased from 3a and 3b to less hydrophilic 4a and 4b, indicated that the suitable hydrophilicity/lipophilicity of a compound was important for the anti-proliferation of cancer cells. An important finding was that MDA-MB-231 was more sensitive to these series of compounds than A549, and 3a, 3b and 5a were three times more active towards MDA-MB-231 than towards A549.
Derived from a selective anticancer cell proliferative agent 5a, 19-acetylated compound 6a (Table 1, entry 9) had no anti-proliferative effect on the two cancer cell lines but became a selective toxic agent to zebrafish, which showed toxicity to zebrafish at 30 μM, suggesting that the action of 6a to zebrafish possibly came from its static inhibition. 19-Acetylated compound 6b (Table 1, entry 10) exhibited its more selective anticancer cell proliferation (>8-fold difference) to MDA-MB-231 than to A549 with CC50 values of 6.7 and 56.5 μM, respectively; moreover, 6b showed its toxicity to zebrafish up to 300 μM, which was considerably less toxic to zebrafish than 19-alcohol compound 5b. It is obvious that the difference of the selectivity and potency of 6a and 6b (ClogP > 5 and molecular weight > 500) was due to their distinct 14-stereochemistry. Compared to mono-acetylated compound 6a, 3,19-diacetylated compound 7a (Table 1, entry 11) was not active to cancer cell proliferation and also lost the toxicity to zebrafish. 3,19-Diacetylated compound 7b (Table 1, entry 12) retained the same level of toxicity to zebrafish at 300 μM as 6b, but 7b became a much weaker anti-proliferative agent to two cancer cell lines than 6b, and interestingly, 7b still possessed the selectivity to MDA-MB-231 over A549 as 6b did. Further studies showed that among 14,19-di-OTBS compounds, only 3-alcohol 8b (Table 1, entry 14) showed weak anticancer cell proliferative activity, whereas 3-alcohol 8a (Table 1, entry 13), 3-acetylated compounds 9a and 9b (Table 1, entries 15 and 16) and 3,14,19-tri-OTBS compounds 10a and 10b (Table 1, entries 17 and 18) were not active in anticancer cell proliferation; moreover, all the 14,19-di-OTBS compounds 8a, 8b, 9a, 9b, 10a and 10b did not exhibit any toxicity to zebrafish.
In contrast to compounds 9a and 9b, more hydrophilic compounds 11a and 11b (ClogP > 5 and molecular weight > 500) (Table 1, entries 19 and 20) bearing 3-OAc, not only exhibited potent anti-proliferative effects on MDA-MB-231 and A549 with CC50 values of 4.7, 9.8 and 4.3, 11.3 μM, respectively, but also showed very strong toxicity to zebrafish. However, the changes of the hydrophilicity from 10a and 10b to 12a and 12b (Table 1, entries 21 and 22), which contained 3-OTBS, did not affect their anti-proliferative effect on MDA-MB-231 and A549 and toxicity to zebrafish, i.e., they were not active to two cancer cell lines and not toxic to zebrafish embryo development. By the comparison of 5a with 11a, it can be inferred that 3-OAc heavily enhanced the toxicity to zebrafish even though 3-OAc did not considerably change from 5b to 11b. It is concluded from these results that 3,14,19-substituted groups, which contributed to the lipophilicity of the compound, together with the stereochemistry of 14-OTBS determined the anti-proliferative activity of a compound against cancer cells and its toxicity to zebrafish. Moreover, the selectivity of a compound's anticancer cell proliferative activity was MDA-MB-231 over A549. Notably, groups of 14-OTBS and 19-OH provided a higher chance to exhibit toxicity to zebrafish (5b, 11a and 11b).
How acetylation at 14-position and 3-ketone affected a compound's anti-proliferative effect on the cancer cell lines and toxicity to zebrafish were also explored. Both the 14-acetylated analogues 13a and 13b (Table 1, entries 23 and 24) were active against the anti-proliferation of two cancer cell lines and toxic to zebrafish development, and 13b of 14β-isomer was more active to two cancer cell lines and more toxic to zebrafish than 13a of 14α-isomer. Compared with 13a and 13b, the removal of the 3,19-acetonylidene group made more hydrophilic compounds 14a and 14b (Table 1, entries 25 and 26) were two times less active to two cancer cell lines than 13a and 13b, and 14a showed slightly better activity than 14b in anticancer cell proliferation. Moreover, 14a exhibited considerably weaker toxicity to zebrafish than 13a, and 14b totally lost the toxicity to zebrafish, indicating that herein, the hydrophilicity decreased the toxicity to zebrafish, and the 14β-OAc isomer of 14b possessed selective anticancer cell proliferative activities to MDA-MB-231 and A549. Upon the introduction of TBS into 19-position, 3-alcohol compounds 15a and 15b (ClogP > 5 and molecular weight > 500) with 14-OAc and 19-OTBS (Table 1, entries 27 and 28) and 3-ketone compounds 16a and 16b (ClogP > 5 and molecular weight > 500) with 14-OAc and 19-OTBS (Table 1, entries 29 and 30) showed the strongest anti-proliferative activities towards two cancer cell lines and also exhibited the strongest toxicities to zebrafish, suggesting that their anti-proliferative activity towards two cancer cell lines and toxicity to zebrafish were correlated and the combination of 14-OAc and 19-OTBS was crucial in playing anti-proliferation activity and toxicity. It is valuable that the toxicities to zebrafish of 14β-isomers of 15b and 16b were stronger than those of 14α-isomers of 15a and 16a, but the transformation of 3-alcohol 15a and 15b to 3-ketone 16a and 16b had no influence on the activity and toxicity. Moreover, by the comparison of 15a/15b with 6a/6b and 11a/11b, it was found that different positions of OTBS and OAc afforded different anti-proliferative activity and zebrafish toxicity.
After the removal of 19-TBS from 3-ketones 16a and 16b, 3-ketones 17a and 17b (Table 1, entries 31 and 32) with 14-OAc–19-OH, which efficiently followed the “Rule of Five”, were selective agents against cell proliferation of MDA-MB-231 and A549, and importantly, 17a is the sole compound in this study that is more active to A549 than to MDA-MB-231. The transformation of 3-alcohol 14a (Table 1, entry 25) to 3-ketone 17a did not change the anticancer cell proliferative activity towards MDA-MB-231 but increased the anticancer cell proliferative activity towards A549 by more than 2 times, and 17a was a safe compound for zebrafish; in the other direction, the transformation from alcohol 14b to ketone 17b of 14β-isomers (Table 1, entries 26 and 32) almost did not change the activity and toxicity. Compared to 19-OTBS 15a and 15b, 19-OAc compounds 18a and 18b with 14-OAc and 3-OH (Table 1, entries 33 and 34), which obey the “Rule of Five”, selectively inhibited the cell proliferation of MDA-MB-231 and A549 but did not exhibit toxicity to zebrafish; it is interesting that 14β-isomer of 18b was two times more active to cancer cell proliferation than 14α-isomer of 18a. 3-Ketone compounds 19a and 19b with 14,19-di-OAc (Table 1, entries 35 and 36) showed very close anticancer cell proliferative activities to their corresponding 3-alcohol compounds 18a and 18b; however, unlike 18a and 18b, 19a exhibited toxicity at 100 μM to zebrafish and 19b exhibited toxicity at 30 μM to zebrafish and killed zebrafish at 100 μM, indicating that 3-ketone deeply affected the toxicity to zebrafish and less hydrophilicity increased the toxicity. After the hydrolysis of 14,19-di-OAc from 19a and 19b, 3-ketones 20a and 20b (Table 1, entries 37 and 38) bearing 14,19-di-OH became safe for zebrafish similar to 1 and 2 (Table 1, entries 1 and 2) but lost anticancer cell proliferative activity. The introduction of TBS to provide 21a and 21b generated the biggest difference of activity and toxicity between 14α-isomer 21a and 14β-isomer 21b: even though 3-ketone 21a bearing 14α-OH–19-OTBS (Table 1, entry 39) was not active against cancer cell proliferation and safe to zebrafish, ketone 21b bearing 14β-OH–19-OTBS (Table 1, entry 40) exhibited very strong anticancer cell proliferative activity and also strong toxicity to zebrafish. These data indicated that 14-stereochemistry, 19-OTBS and the combination of 14-OAc and 19-OTBS exerted important roles in the anti-proliferative activity of two cancer cell lines and toxicity to zebrafish.
Unlike its 3-alcohol mother compound 6a (Table 1, entry 9), which was a selective toxic agent to zebrafish, 3-ketone 22a (ClogP > 5 and molecular weight > 500) bearing 14-OTBS and 19-OAc (Table 1, entry 41) was considerably less toxic to zebrafish than 6a but 22a was a potent anticancer proliferation agent with IC50 values of 3.5 μM and 8.9 μM to MDA-MB-231 and A549, respectively, reflecting that 3-alcohol (6a) and 3-ketone (22a) determined the selectivity towards anti-proliferative activity of two cancer cells and toxicity to zebrafish. Unlike 6b (Table 1, entry 10), 3-ketone 22b (Table 1, entry 42) was the most toxic compound to zebrafish; however, as 6b, 22b also showed its selective anticancer cell proliferative activity (>5-fold difference) to MDA-MB-231 over A549 with CC50 values of 3.6 and 21.6 μM, respectively. Compared to 19a, 19b, 22a and 22b, more polar 3-keto-14-OH–19-OAc 23a and 23b (Table 1, entries 43 and 44) exhibited reduced toxicity to zebrafish and their anticancer cell proliferative effects were also less, but 14α-isomer 23a was more active than 14β-isomer 23b against cancer cell proliferation, suggesting that the substitution and stereochemistry at 14-position play a key role in the anti-proliferative activity and toxicity. Similar to its mother 3-alcohol 8a (Table 1, entry 13), 3-keto-14α,19-di-OTBS 24a (Table 1, entry 45) was not active against cancer cell proliferation and safe to zebrafish; moreover, 3-keto-14β,19-di-OTBS 24b (Table 1, entry 46) was not toxic to zebrafish as 3-alcohol 8b (Table 1, entry 14), but 24b showed only very weak anti-proliferative activity to MDA-MB-231. Furthermore, the removal of 19-OTBS from 24a and 24b sharply increased the toxicity to zebrafish and the anticancer cell proliferative activities of 25a and 25b (5.2796 of ClogP, Table 1, entries 47 and 48) with 14-OTBS substitution, as well. In contrast to 3-ketone 25a (Table 1, entry 47) with 3-alcohol 5a (Table 1, entry 7), 3-keto group did not affect A549 cancer cells but was to reduce anti-proliferative activity towards MDA-MB-231 and a cofactor to enhance the toxicity to zebrafish; in addition, compared with alcohol 5b, 3-ketone 25b (Table 1, entry 48) showed similar anticancer cell proliferative activity to A549 and the same level of toxicity to zebrafish but was more active against the cell proliferation of MDA-MB-231, indicating that MDA-MB-231 was more sensitive than A549 to the transformation of 3-OH to 3-ketone, and different 14-stereochemistry produced opposite changes in the activity and toxicity. These data suggest that the selectivity of one compound might be partly derived from the property of the substitution.
Conclusions
The small library in this paper was diversified by 14α- and 14β-isomers, different substituents and mono- or multi-substitutions at 3,14,19-positions, and the transformation of 3-alcohol to 3-ketone. Because our goal in this study was to discover in vitro anticancer cell proliferative compounds of andrographolide derivatives and summarize the SAR of the small library, we also used an in vivo zebrafish model to analyze the toxicity of these andrographolide derivatives and conclude the structure–toxicity relationship of the small library.
Compared to 1 and 2, the modifications at 3-,14-,19-positions changed the hydrophilicity of a compound, which was expected to affect activity and toxicity via its cell permeability to some extent. Compounds 4a/4b, 7a/7b, 9a/9b, 10a/10b, 13a/13b, 16a/16b, 19a/19b, 22a/22b and 24a/24b have higher lipophilicity among the compounds studied; however, most of them were not much active and/or much toxic except 13a/13b, 16a/16b and 22b. These data suggest that the anti-proliferative activity towards cancer cells and toxicity to zebrafish were only in part derived from the permeability, and optimal hydrophilicity/lipophilicity was possibly crucial for anti-cancer cell proliferative activity and zebrafish toxicity.
It is interesting and important that some modifications at 1 led to the selectivity of anticancer cell proliferative activity or zebrafish toxicity. Compound 6a was a selective toxic agent to zebrafish, and did not exhibit anticancer cell proliferative activity up to 300 μM; however, compound 6b showed more selective anti-proliferative effects towards breast cancer MDA-MB-231 cells (>8-fold difference) than non-small cell lung cancer cell A549 but no toxicity to zebrafish up to 100 μM. Compound 17a was not toxic to zebrafish; importantly and interestingly, 17a showed more selective anti-proliferative activity to A549 than to MDA-MB-231 even though all the other compounds studied in this paper were more active against the cell proliferation of MDA-MB-231 than A549. It was discovered that 1, 2, 5a, 8b, 14b, 17a, 17b, 18a, 18b and 23a were selective compounds against the cancer cell proliferation of MDA-MB-231 and A549. In addition, 3a, 3b, 14a and 22a showed low CC50 values to both A549 and MDA-MB-231 cells (lower than 20 μM), and they had no toxicity to zebrafish at concentrations up to 100 μM.
It was unveiled that the selectivity difference between the anti-proliferative activity of cancer cells and toxicity to zebrafish of some compounds only originated from 14α- and 14β-stereochemistry. The 14α-isomers of 5a and 22a showed good anticancer cell proliferative effects without toxicity or very low toxicity to zebrafish, but their 14β-isomers of 5b and 22b not only exhibited good anticancer cell proliferative activity but also expressed very high toxicity to zebrafish. The 14α-isomer of 6a was very toxic to zebrafish but did not show anticancer cell proliferative activity; however, 14β-isomer of 6b exhibited good anticancer cell proliferative activity with low toxicity to zebrafish. Although the compound 21a was safe to zebrafish and was not active to cancer cell proliferation, 14β-isomer of 21b was an excellent anticancer cell proliferation inhibitor and it was also very toxic to zebrafish.
The transformation between 3-alcohol and 3-ketone sometimes changed the activity, the toxicity and the selectivity. 3-Alcohol 6a appeared to be a selective agent to zebrafish, but its corresponding 3-ketone 22a was a very potent inhibitor to cancer cell proliferation with very low toxicity to zebrafish. On the other hand, 3-ketones 19a and 19b showed similar anticancer cell proliferative activities to their corresponding 3-alcohols 18a and 18b but ketones 19a and 19b were strongly toxic to zebrafish embryo development. 3-Ketone 25a became a toxic compound to zebrafish and less active inhibitor to the cell proliferation of MDA-MB-231, while 3-alcohol 5a was a selective agent to cancer cell proliferation. In addition, the transformation of 3-alcohol 5a to 3-OAc 11a made 11a a toxic agent to zebrafish; unlike very active and toxic 19-OTBS compounds 16a and 16b, 19-OH compounds 17a and 17b possessed good anticancer cell proliferative activity but were safe to zebrafish embryo development.
From the analysis of the most active and also most toxic compounds 5b, 11a/11b, 13a/13b, 15a/15b, 16a/16b, 19b, 21b, 22b, and 25b, it could be concluded that 14β-isomers were possibly more active to cancer cell proliferation and toxic to zebrafish as well, and the dual substitutions of TBS and Ac at 14 and 19-positions (16b and 22b) were more feasible to bring about toxicity to zebrafish development. On the other hand, substitutions of OH, OTBS and OAc at different positions at 3, 14 and 19 positions afforded different anti-proliferative activity of cancer cells and toxicity to zebrafish, as observed by the comparison of the activities and toxicities of 6a/6b, 11a/11b and 15a/15b.
Although the compounds studied in this paper are not the drug candidates from the point of drug discovery and development (calculated ClogP values are listed in Table 1), 5a, 14a, 14b, 17a, 17b, 18a, 18b and 22a could be leads or potential candidates for further anti-cancer drug discovery and development of andrographolide derivatives. In particular, 6b was a selective hit against MDA-MB-231 proliferation and could be developed into a selective anti-proliferative drug candidate to MDA-MB-231; 17a could be a lead to discover the selective anti-proliferative inhibitors of A549; 6a would be studied in the future as a possible selective static inhibitor to zebrafish development; and modification of 15b, 16b, 19b, 21b, 22b and 25b by truncating their toxicity (“detoxification”) and increasing their anticancer cell proliferative activity should be a possible and rational strategy to drug discovery. Targeting on drug discovery and development of anticancer agents, one of our future missions is to focus on the replacement of TBS and/or Ac with more stable and suitable hydrophilic groups in view of druggability and Lipinski's “Rule of Five”. In addition, the elucidation of the toxic mechanisms of these compounds against zebrafish embryo development and the discovery of drug with excellent static inhibitory agents (possibly like 6a) to zebrafish embryo development are also promising goals in our future research.
In summary, our SAR data indicated that the stereochemistry of 14-position of 1 and specific single or suitably combined modifications at 3-, 14- and 19-positions of 1 affected not only in vitro anticancer cell proliferative activity to MDA-MB-231 and A549 and the in vivo toxicity to zebrafish, but also the selectivity between the in vitro anti-proliferative activity, and the in vivo toxicity, and different cancer cell lines. Overall, some potential leads or hits for further anti-cancer drug development and a possible static inhibitor to zebrafish embryo development were discovered from this research; moreover, the structure–toxicity relationships in zebrafish suggested that the direct and/or indirect toxicity of an active compound with andrographolide pharmacophore should be given attention.
Experimental
Materials and equipment
Unless otherwise stated, materials were obtained from commercial suppliers and used without further purification. 1H and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer at 400 and 101 MHz, respectively, in an indicated deuterated solvent. Coupling constants (J) are expressed in hertz (Hz). Chemical shifts (δ) of NMR are reported in parts per million (ppm) units relative to the solvent. The high resolution of MS (HRMS) was recorded on an Applied Biosystems Q-STAR Elite ESI-LC-MS/MS mass spectrometer. Melting points were measured using a YRT-3 melting point apparatus (Shanghai, China) and were uncorrected.
Preparation of the compounds 4a11 and 4b
Under N2 atmosphere, 10.0 g (25.6 mmol) of compound 3a5 or 3b5 and 6.0 ml (51.3 mmol) of 2,6-lutidine were dissolved in 60.0 ml anhydrous dichloromethane. The solution was cooled to 0 °C and then 8.8 ml (38.5 mmol) of TBSOTf was added dropwise over 5 min. The reaction mixture was stirred for 0.5 h at 0 °C and treated with ethyl acetate and sat. NaHCO3 solution after the reaction was complete. The organic phase was washed with brine for 6 times, dried over anhydrous Na2SO4, filtered and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/15) to provide compound 4a or 4b.
3,19-Acetonylidene-14α-tert-butyldimethylsilyloxy-andrographolide (4a). 86% yield, white solid, mp 128.6–129.5 °C. 1H NMR (400 MHz, C6D6) δ 7.00 (1H, ddd, J = 7.3, 5.8, 2.0 Hz, 12-H), 4.86 (1H, s, 17α-H), 4.69 (1H, s, 17β-H), 4.49–4.43 (1H, m, 14β-H), 3.85 (1H, d, J = 11.9 Hz, 19α-H), 3.81 (1H, dd, J = 9.7, 6.8 Hz, 15α-H), 3.70 (1H, dd, J = 9.6, 3.5 Hz, 15β-H), 3.50 (1H, dd, J = 7.4, 3.0 Hz, 3β-H), 3.10 (1H, d, J = 11.6 Hz, 19β-H), 2.58 (1H, ddd, J = 17.8, 10.9, 7.0 Hz, 11α-H), 2.25–2.14 (2H, m, 9β-H and 11β-H), 1.98–1.88 (1H, m, 7α-H), 1.81–1.69 (1H, m, 7β-H), 1.69–1.57 (2H, m, 1α-H and 2α-H), 1.51 (1H, d, J = 11.8 Hz, 1β-H), 1.44 (3H, s, CCH3), 1.39 (3H, s, CCH3), 1.37–1.29 (1H, m, 2β-H), 1.11 (3H, s, 18-H), 1.10–1.02 (1H, m, 6α-H), 1.02 (3H, s, 20-H), 1.00–0.89 (2H, m, 5β-H and 6β-H), 0.87 (9H, s, SiC(CH3)3), −0.00 (3H, s, SiCH3), −0.17 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 169.1 (16-C), 147.5 (12-C), 147.4 (8-C), 128.2 (13-C), 109.9 (17-C), 99.6 (C(CH3)2), 75.4 (3-C), 73.4 (15-C), 67.4 (14-C), 64.4 (19-C), 56.2 (9-C), 51.3 (5-C), 38.4 (4-C), 38.3 (10-C), 37.8 (7-C), 34.4 (1-C), 26.6 (2-C), 26.2 (6-C), 25.8 (SiC(CH3)3), 25.4 (CCH3), 25.0 (CCH3), 24.9 (11-C), 23.3 (18-C), 17.9 (SiC(CH3)3), 17.1 (20-C), −4.3 (SiCH3), −4.8 (SiCH3); HRMS (ESI) m/z 527.3190 [M + Na]+, calculated for C29H48O5SiNa, 527.3169.
3,19-Acetonylidene-14β-tert-butyldimethylsilyloxy-androgra-pholide (4b). 70% yield, white solid, mp 158.3–159.2 °C. 1H NMR (400 MHz, C6D6) δ 6.94 (1H, ddd, J = 8.2, 4.7, 2.0 Hz, 12-H), 4.88 (1H, d, J = 1.2 Hz, 17α-H), 4.58–4.53 (1H, m, 14α-H), 4.43 (1H, d, J = 0.9 Hz, 17β-H), 3.86 (1H, d, J = 11.5 Hz, 19α-H), 3.76 (1H, dd, J = 9.7, 6.3 Hz, 15α-H), 3.72 (1H, dd, J = 9.7, 3.5 Hz, 15β-H), 3.47 (1H, dd, J = 7.3, 3.1 Hz, 3β-H), 3.11 (1H, d, J = 11.5 Hz, 19β-H), 2.51–2.31 (2H, m, 9β-H and 11α-H), 2.21 (1H, ddd, J = 12.8, 3.9, 2.4 Hz, 11β-H), 1.94–1.82 (1H, m, 7α-H), 1.74 (1H, td, J = 13.0, 5.0 Hz, 7β-H), 1.63–1.52 (2H, m, 1α-H and 2α-H), 1.49 (1H, dd, J = 10.1, 1.1 Hz, 2β-H), 1.43 (3H, s, CCH3), 1.40–1.33 (4H, m, CCH3 and 1β-H), 1.10 (3H, s, 18-H), 1.08–1.02 (2H, m, 6α-H and 5β-H), 1.02 (3H, s, 20-H), 0.90 (1H, dd, J = 12.8, 2.6 Hz, 6β-H), 0.86 (9H, s, SiC(CH3)3), −0.00 (3H, s, SiCH3), −0.16 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 169.1 (16-C), 148.5 (12-C), 147.4 (8-C), 128.6 (13-C), 108.3 (17-C), 99.6 (C(CH3)2), 75.3 (3-C), 73.4 (15-C), 67.7 (14-C), 64.4 (19-C), 56.1 (9-C), 51.1 (5-C), 38.4 (4-C), 38.3 (10-C), 37.8 (7-C), 34.1 (1-C), 26.5 (2-C), 26.1 (6-C), 25.7 (SiC(CH3)3), 25.6 (CCH3), 25.4 (CCH3), 24.8 (11-C), 23.4 (18-C), 18.0 (SiC(CH3)3), 17.1 (20-C), −4.2 (SiCH3), −4.8 (SiCH3); HRMS (ESI) m/z 527.3185 [M + Na]+, calculated for C29H48O5SiNa, 527.3169.
Preparation of the compounds 5a12 and 5b
10.0 g (19.8 mmol) of compound 4a or 4b was dissolved in 20.0 ml of methanol. The solution was treated with 0.38 g (2.0 mmol) of p-TSA at 0 °C for 0.5 h. Diluted by ethyl acetate and washed with sat. NaHCO3 solution and brine; the organic phase was dried over anhydrous Na2SO4, filtered, and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/1) to afford compound 5a or 5b.
14α-tert-Butyldimethylsilyloxy-andrographolide (5a). 98% yield, white solid, mp 116.3–117.8 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.60 (1H, td, J = 6.2, 1.4 Hz, 12-H), 5.19 (1H, d, J = 6.1 Hz, 14β-H), 5.05 (1H, d, J = 4.8 Hz, 3α-OH), 4.79 (1H, s, 17α-H), 4.50 (1H, s, 17β-H), 4.46 (1H, dd, J = 10.0, 5.9 Hz, 15α-H), 4.11 (1H, dd, J = 7.5, 2.8 Hz, 19-OH), 4.00 (1H, dd, J = 10.0, 2.3 Hz, 15β-H), 3.83 (1H, dd, J = 10.9, 2.7 Hz, 19α-H), 3.25 (2H, td, J = 9.9, 8.8, 6.5 Hz, 3β-H and 19β-H), 2.44 (2H, t, J = 7.0 Hz, 11-H), 2.32 (1H, dt, J = 13.0, 3.1 Hz, 9β-H), 1.95 (2H, q, J = 8.0, 7.0 Hz, 7-H), 1.78–1.69 (1H, m, 2α-H), 1.68–1.57 (3H, m, 1-H and 2β-H), 1.35 (1H, qd, J = 12.9, 3.9 Hz, 6α-H), 1.28–1.17 (2H, m, 5β-H and 6β-H), 1.08 (3H, s, 18-H), 0.86 (9H, s, SiC(CH3)3), 0.65 (3H, s, 20-H), 0.15 (3H, s, SiCH3), 0.12 (3H, s, SiCH3); HRMS (ESI) m/z 487.2867 [M + Na]+, calculated for C26H44O5SiNa, 487.2856.
14β-tert-Butyl-dimethylsilyloxy-andrographolide (5b). 95% yield, white solid, mp 153.7–154.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.57 (1H, ddd, J = 7.9, 4.2, 1.4 Hz, 12-H), 5.23 (1H, d, J = 5.7 Hz, 14α-H), 5.06 (1H, d, J = 4.3 Hz, 3α-OH), 4.79 (1H, s, 17α-H), 4.48 (1H, dd, J = 9.9, 6.1 Hz, 15α-H), 4.32 (1H, s, 17β-H), 4.16–4.06 (1H, m, 19-OH), 3.99 (1H, dd, J = 9.9, 2.4 Hz, 15β-H), 3.83 (1H, d, J = 10.8 Hz, 19α-H), 3.24 (2H, ddd, J = 15.3, 10.0, 5.6 Hz, 3β-H and 19β-H), 2.49–2.27 (3H, m, 9β-H and 11-H), 1.96 (2H, qd, J = 13.0, 12.0, 3.5 Hz, 7-H), 1.80–1.71 (1H, m, 2α-H), 1.70–1.54 (3H, m, 1-H and 2β-H), 1.43–1.18 (3H, m, 5β-H and 6-H), 1.08 (3H, s, 18-H), 0.87 (9H, s, SiC(CH3)3), 0.65 (3H, s, 20-H), 0.16 (3H, s, SiCH3), 0.11 (3H, s, SiCH3); 13C NMR (101 MHz, DMSO-d6) δ 169.3 (16-C), 147.9 (12-C), 147.3 (8-C), 128.1 (13-C), 107.9 (17-C), 78.3 (3-C), 73.8 (15-C), 66.7 (14-C), 62.6 (19-C), 55.1 (9-C), 54.3 (5-C), 42.2 (4-C), 38.4 (10-C), 37.4 (7-C), 36.7 (1-C), 27.8 (2-C), 25.5 (SiC(CH3)3), 24.9 (6-C), 24.0 (11-C), 23.0 (18-C), 17.5 (SiC(CH3)3), 14.9 (20-C), −4.4 (SiCH3), −5.0 (SiCH3); HRMS (ESI) m/z 487.2868 [M + Na]+, calculated for C26H44O5SiNa, 487.2856.
Preparation of the compounds 6a and 6b
At room temperature, 3.0 g (6.5 mmol) of compound 5a or 5b and 4.04 ml (29.1 mmol) of TEA were dissolved in 20.0 ml ethyl acetate and then 1.61 ml (22.6 mmol) of AcCl was added dropwise over 2 min. The reaction mixture was stirred for 2 h at room temperature and treated with ethyl acetate and sat. NaHCO3 solution after the reaction was complete. The organic phase was washed with brine, dried over anhydrous Na2SO4, filtered and then concentrated under reduced pressure. Purification from the residue to obtain compound 6a or 6b was conducted by silica gel column chromatography (ethyl acetate/petroleum ether 1/6).
14α-tert-Butyldimethyl-silyloxy-19-acetoxy-andrographolide (6a). 65% yield, white solid, mp 129.7–130.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.64–6.57 (1H, m, 12-H), 5.20 (1H, d, J = 5.7 Hz, 14β-H), 4.81 (1H, s, 17α-H), 4.72 (1H, d, J = 4.7 Hz, 3α-OH), 4.51 (1H, s, 17β-H), 4.47 (1H, dd, J = 10.0, 5.9 Hz, 15α-H), 4.17–4.07 (2H, m, 19-H), 4.01 (1H, dd, J = 10.0, 2.3 Hz, 15β-H), 3.18 (1H, dt, J = 10.4, 5.0 Hz, 3β-H), 2.46 (2H, t, J = 6.5 Hz, 9β-H and 11α-H), 2.33 (1H, dt, J = 12.4, 2.4 Hz, 11β-H), 1.99 (1H, s, 7α-H), 1.96 (3H, s, CH3CO), 1.96–1.89 (1H, m, 7β-H), 1.81 (1H, d, J = 11.2 Hz, 2α-H), 1.67 (1H, d, J = 13.0 Hz, 2β-H), 1.63–1.50 (2H, m, 1-H), 1.44 (1H, td, J = 13.3, 4.2 Hz, 6α-H), 1.34–1.21 (2H, m, 5β-H and 6β-H), 1.04 (3H, s, 18-H), 0.87 (9H, s, SiC(CH3)3), 0.68 (3H, s, 20-H), 0.15 (3H, s, SiCH3), 0.12 (3H, s, SiCH3); 13C NMR (101 MHz, DMSO-d6) δ 170.4 (16-C), 169.4 (CH3CO), 147.8 (12-C), 147.4 (8-C), 127.6 (13-C), 108.4 (17-C), 76.5 (3-C), 73.9 (15-C), 66.4 (14-C), 65.1 (19-C), 55.3 (9-C), 53.9 (5-C), 41.6 (4-C), 38.6 (10-C), 37.5 (7-C), 36.8 (1-C), 27.5 (2-C), 25.6 (SiC(CH3)3), 24.6 (6-C), 24.2 (11-C), 22.8 (18-C), 20.9 (CH3CO), 17.5 (SiC(CH3)3), 14.2 (20-C), −4.5 (SiCH3), −5.0 (SiCH3); HRMS (ESI) m/z 529.2972 [M + Na]+, calculated for C28H46O6SiNa, 529.2961.
14β-tert-Butyldimethylsilyloxy-19-acetoxy-andrographolide (6b). 58% yield, white solid, mp 149.8–150.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.62–6.54 (1H, m, 12-H), 5.24 (1H, d, J = 5.5 Hz, 14α-H), 4.80 (1H, s, 17α-H), 4.74 (1H, d, J = 4.6 Hz, 3α-OH), 4.48 (1H, dd, J = 9.9, 6.1 Hz, 15α-H), 4.33 (1H, s, 17β-H), 4.11 (2H, q, J = 11.7 Hz, 19-H), 3.99 (1H, dd, J = 9.9, 2.4 Hz, 15β-H), 3.17 (1H, dt, J = 10.8, 5.1 Hz, 3β-H), 2.49–2.28 (3H, m, 9β-H, 11-H), 2.06–1.99 (1H, m, 7α-H), 1.96 (3H, s, CH3CO), 1.92 (1H, dd, J = 13.7, 4.1 Hz, 7β-H), 1.86–1.77 (1H, m, 2α-H), 1.65–1.50 (3H, m, 1-H and 2β-H), 1.47 (1H, dd, J = 13.1, 3.6 Hz, 6α-H), 1.36–1.21 (2H, m, 5β-H and 6β-H), 1.03 (3H, s, 18-H), 0.86 (9H, s, SiC(CH3)3), 0.68 (3H, s, 20-H), 0.16 (3H, s, SiCH3), 0.11 (3H, s, SiCH3); 13C NMR (101 MHz, DMSO-d6) δ 170.4 (16-C), 169.3 (CH3CO), 147.9 (12-C), 147.3 (8-C), 128.1 (13-C), 107.9 (17-C), 76.5 (3-C), 73.9 (15-C), 66.7 (14-C), 65.0 (19-C), 55.2 (9-C), 53.9 (5-C), 41.6 (4-C), 38.6 (10-C), 37.6 (7-C), 36.8 (1-C), 27.5 (2-C), 26.4 (6-C), 25.5 (SiC(CH3)3), 24.8 (11-C), 22.8 (18-C), 20.9 (CH3CO), 17.5 (SiC(CH3)3), 14.2 (20-C), −4.4 (SiCH3), −5.0 (SiCH3); HRMS (ESI) m/z 529.2956 [M + Na]+, calculated for C28H46O6SiNa, 529.2961.
Preparation of the compounds 7a and 7b
0.5 g (1.0 mmol) of compound 6a or 6b and 0.013 g (0.1 mmol) of anhydrous ZnCl2 were dissolved in 10.0 ml Ac2O. The solution was heated to 50 °C and stirred for 1 h to complete the reaction. The resulting mixture was treated with ethyl acetate and sat. NaHCO3 solution after it was cooled to room temperature. The organic phase was washed with brine, dried over anhydrous Na2SO4, filtered, concentrated, and then purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/8) to obtain compound 7a or 7b.
14α-tert-Butyldimethylsilyloxy-3,19-di-acetoxy-andrographolide (7a). 88% yield, white solid, mp 186.2–186.8 °C. 1H NMR (400 MHz, C6D6) δ 6.96 (1H, ddd, J = 7.3, 5.2, 2.0 Hz, H-12), 4.83 (1H, s, 17α-H), 4.71 (1H, dd, J = 12.0, 4.4 Hz, 3β-H), 4.67 (1H, s, 17β-H), 4.57 (1H, d, J = 11.8 Hz, 19α-H), 4.51–4.46 (1H, m, 14β-H), 4.09 (1H, d, J = 11.8 Hz, 19β-H), 3.84 (1H, dd, J = 9.6, 6.6 Hz, 15α-H), 3.71 (1H, dd, J = 9.6, 3.6 Hz, 15β-H), 2.48 (1H, ddd, J = 17.7, 10.9, 7.7 Hz, 11α-H), 2.19 (1H, ddd, J = 12.9, 3.7, 2.3 Hz, 11β-H), 2.10 (1H, d, J = 17.2 Hz, 9β-H), 1.78–1.71 (2H, m, 7-H), 1.71 (3H, s, CH3CO), 1.69 (3H, s, CH3CO), 1.67–1.55 (2H, m, 2-H), 1.51–1.41 (2H, m, 1-H), 1.30 (1H, qd, J = 13.1, 4.1 Hz, 6α-H), 1.01 (3H, s, 18-H), 0.99–0.89 (2H, m, 5β-H and 6β-H), 0.87 (9H, s, SiC(CH3)3), 0.66 (3H, s, 20-H), −0.02 (3H, s, SiCH3), −0.16 (3H, s, SiCH3); 13C NMR (101 MHz, DMSO-d6) δ 170.2 (16-C), 169.9 (CH3CO), 169.4 (CH3CO), 147.4 (12-C), 147.2 (8-C), 127.7 (13-C), 108.8 (17-C), 79.1 (3-C), 73.9 (15-C), 66.4 (14-C), 63.6 (19-C), 54.8 (9-C), 53.8 (5-C), 40.9 (4-C), 38.4 (10-C), 37.1 (7-C), 36.3 (1-C), 25.6 (SiC(CH3)3), 24.2 (2-C), 24.0 (6-C), 23.9 (11-C), 22.3 (18-C), 20.9 (CH3CO), 20.8 (CH3CO), 17.5 (SiC(CH3)3), 14.3 (20-C), −4.5 (SiCH3), −5.0 (SiCH3); HRMS (ESI) m/z 566.3527 [M + NH4]+, calculated for C30H49NO7Si, 566.3513.
14β-tert-Butyldimethylsilyloxy-3,19-diacetoxy-andrographolide (7b). 92% yield, white solid, mp 152.7–153.6 °C. 1H NMR (400 MHz, C6D6) δ 6.93 (1H, ddd, J = 6.7, 4.5, 1.8 Hz, 12-H), 4.84 (1H, s, 17α-H), 4.68 (1H, dd, J = 11.9, 4.5 Hz, 3β-H), 4.58–4.49 (2H, m, 14α-H and 19α-H), 4.37 (1H, s, 17β-H), 4.11 (1H, d, J = 11.8 Hz, 19β-H), 3.80–3.73 (1H, m, 15α-H), 3.72–3.67 (1H, m, 15β-H), 2.41–2.17 (3H, m, 11-H and 9β-H), 1.74 (1H, dd, J = 13.3, 4.4 Hz, 7α-H), 1.69 (6H, d, J = 3.7 Hz, CH3CO and CH3CO), 1.67–1.51 (3H, m, 2-H and 7β-H), 1.50–1.27 (3H, m, 1-H and 6α-H), 1.00 (3H, s, 18-H), 0.92 (2H, d, J = 12.8 Hz, 5β-H and 6β-H), 0.86 (9H, s, SiC(CH3)3), 0.67 (3H, s, 20-H), −0.02 (3H, s, SiCH3), −0.17 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 170.0 (16-C), 169.7 (CH3CO), 169.0 (CH3CO), 147.9 (12-C), 147.0 (8-C), 128.5 (13-C), 108.2 (17-C), 79.4 (3-C), 73.3 (15-C), 67.7 (14-C), 64.6 (19-C), 55.6 (9-C), 54.6 (5-C), 41.5 (4-C), 38.9 (10-C), 38.0 (7-C), 37.0 (1-C), 25.7 (SiC(CH3)3), 25.3 (2-C), 24.8 (6-C), 24.5 (11-C), 22.7 (18-C), 20.73 (CH3CO), 20.65 (CH3CO), 18.0 (SiC(CH3)3), 14.9 (20-C), −4.2 (SiCH3), −4.8 (SiCH3); HRMS (ESI) m/z 571.3062 [M + Na]+, calculated for C30H48O7SiNa, 571.3067.
Preparation of the compounds 8a and 8b
5.0 g (10.8 mmol) of compound 5a or 5b and 4.4 g (64.7 mmol) of imidazole were dissolved in 30.00 ml anhydrous dichloromethane and then 8.14 g (52.9 mmol) of TBSCl in 5.0 ml of anhydrous dichloromethane was added dropwise over 2 min. The reaction mixture was stirred for 1 h at room temperature and treated with ethyl acetate and sat. NaHCO3 solution after the reaction was complete. The organic phase was washed with brine, dried over anhydrous Na2SO4, filtered and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/8) to afford compound 8a or 8b.
14α,19-Di-tert-butyldimethylsilyloxy-andrographolide (8a). 87% yield, white solid, mp 122.5–123.6 °C. 1H NMR (400 MHz, C6D6) δ 6.96 (1H, td, J = 5.3, 2.6 Hz, 12-H), 4.84 (1H, s, 17α-H), 4.67 (1H, s, 17β-H), 4.52–4.46 (1H, m, 14β-H), 4.22 (1H, d, J = 10.0 Hz, 19α-H), 4.01 (1H, d, J = 7.3 Hz, 3α-OH), 3.83 (1H, dd, J = 9.6, 6.6 Hz, 15α-H), 3.71 (1H, dd, J = 9.6, 3.6 Hz, 15β-H), 3.47–3.37 (2H, m, 3β-H and 19β-H), 2.56–2.43 (1H, m, 11α-H), 2.23–2.12 (2H, m, 9β-H and 11β-H), 2.04 (1H, dq, J = 13.4, 3.7 Hz, 7α-H), 1.78 (2H, qd, J = 13.5, 3.5 Hz, 7β-H and 1α-H), 1.61–1.46 (3H, m, 1β-H and 2-H), 1.31 (3H, s, 18-H), 1.16–1.04 (1H, m, 6α-H), 1.00 (2H, td, J = 13.3, 12.8, 3.4 Hz, 5β-H and 6β-H), 0.92 (9H, s, SiC(CH3)3), 0.87 (9H, s, SiC(CH3)3), 0.67 (3H, s, 20-H), 0.02 (3H, s, SiCH3), 0.01 (3H, s, SiCH3), 0.00 (3H, s, SiCH3), −0.16 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 169.0 (16-C), 147.3 (12-C), 147.2 (8-C), 128.2 (13-C), 109.7 (17-C), 79.9 (3-C), 73.3 (15-C), 67.5 (14-C), 65.6 (19-C), 56.1 (9-C), 55.1 (5-C), 43.0 (4-C), 39.0 (7-C), 38.0 (10-C), 37.7 (1-C), 29.3 (2-C), 26.0 (SiC(CH3)3), 25.8 (SiC(CH3)3), 24.7 (6-C), 24.2 (11-C), 23.4 (18-C), 18.3 (SiC(CH3)3), 18.0 (SiC(CH3)3), 15.8 (20-C), −4.3 (SiCH3), −4.7 (SiCH3), −5.71 (SiCH3), −5.72 (SiCH3); HRMS (ESI) m/z 579.3910 [M + H]+, calculated for C32H59O5Si2, 579.3901.
14β,19-Di-tert-butyl-dimethylsilyloxy-andrographolide (8b). 75% yield, white solid, mp 146.5–147.3 °C. 1H NMR (400 MHz, C6D6) δ 6.92 (1H, ddd, J = 8.6, 4.2, 2.0 Hz, 12-H), 4.86 (1H, s, 17α-H), 4.54 (1H, dd, J = 4.2, 2.0 Hz, 14α-H), 4.38 (1H, s, 17β-H), 4.20 (1H, d, J = 10.0 Hz, 19α-H), 3.90 (1H, d, J = 7.2 Hz, 3α-OH), 3.76 (1H, dd, J = 9.6, 6.4 Hz, 15α-H), 3.70 (1H, dd, J = 9.6, 3.6 Hz, 15β-H), 3.47–3.33 (2H, m, 3β-H and 19β-H), 2.48–2.37 (1H, m, 11α-H), 2.36–2.19 (2H, m, 9β-H and 11β-H), 1.98 (1H, dq, J = 11.2, 3.7 Hz, 7α-H), 1.83–1.68 (2H, m, 7β-H and 2α-H), 1.66–1.57 (1H, m, 2β-H), 1.51 (2H, ddd, J = 22.3, 11.8, 6.4 Hz, 1-H), 1.31 (3H, s, 18-H), 1.15 (1H, qd, J = 13.0, 4.2 Hz, 6α-H), 0.99–0.93 (2H, m, 5β-H and 6β-H), 0.92 (9H, s, SiC(CH3)3), 0.87 (9H, s, SiC(CH3)3), 0.68 (3H, s, 20-H), 0.05–0.05 (9H, m, SiCH3 and Si(CH3)2), −0.16 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 169.0 (16-C), 148.3 (12-C), 147.3 (8-C), 128.4 (13-C), 108.0 (17-C), 79.7 (3-C), 73.3 (15-C), 67.8 (14-C), 65.5 (19-C), 55.9 (9-C), 54.9 (5-C), 43.0 (4-C), 39.0 (10-C), 38.0 (7-C), 37.5 (1-C), 29.2 (2-C), 26.0 (SiC(CH3)3), 25.7 (SiC(CH3)3), 25.4 (6-C), 24.3 (11-C), 23.4 (18-C), 18.3 (SiC(CH3)3), 18.0 (SiC(CH3)3), 15.7 (20-C), −4.2 (SiCH3), −4.8 (SiCH3), −5.7 (Si(CH3)2); HRMS (ESI) m/z 579.3894 [M + H]+, calculated for C32H59O5Si2, 579.3901.
Preparation of the compounds 9a and 9b
1.5 g (2.6 mmol) of compound 8a or 8b and 0.32 g (0.26 mmol) of DMAP were dissolved in 10.0 ml ethyl acetate and then 0.29 ml (3.1 mmol) of Ac2O was added dropwise for 1 min. The reaction mixture was stirred for 24 h at room temperature and treated with ethyl acetate and sat. NaHCO3 solution after the reaction was complete. The organic phase was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated in vacuum. Purification by silica gel column chromatography (ethyl acetate/petroleum ether 1/10) obtained compound 9a or 9b.
3-Acetoxy-14α,19-di-tert-butyldimethylsilyloxy-andrographolide (9a). 89% yield, white solid, mp 127.1–127.8 °C. 1H NMR (400 MHz, C6D6) δ 7.02 (1H, ddd, J = 7.3, 5.6, 2.0 Hz, 12-H), 4.85 (1H, s, 17α-H), 4.80 (1H, dd, J = 11.5, 4.9 Hz, 3β-H), 4.67 (1H, s, 17β-H), 4.52 (1H, dq, J = 4.5, 1.6 Hz, 14β-H), 3.90 (1H, d, J = 10.4 Hz, 19α-H), 3.84 (1H, dd, J = 9.6, 6.5 Hz, 15α-H), 3.77–3.69 (2H, m, 15β-H and 19β-H), 2.53 (1H, ddd, J = 18.5, 11.4, 7.3 Hz, 11α-H), 2.28 (1H, dt, J = 12.3, 2.5 Hz, 9β-H), 2.20 (1H, dt, J = 17.3, 3.7 Hz, 11β-H), 1.83–1.78 (1H, m, 7α-H), 1.77 (3H, s, CH3CO), 1.77–1.63 (4H, m, 7β-H, 2-H and 1α-H), 1.60–1.51 (2H, m, 1β-H and 6α-H), 1.07 (1H, dd, J = 9.8, 3.4 Hz, 5β-H), 1.04 (3H, s, 18-H), 1.02 (1H, d, J = 5.9 Hz, 6β-H), 0.97 (9H, s, SiC(CH3)3), 0.88 (3H, s, 20-H), 0.85 (9H, s, SiC(CH3)3), 0.06 (6H, d, J = 5.6 Hz, Si(CH3)2), −0.00 (3H, s, SiCH3), −0.15 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 170.0 (16-C), 169.0 (CH3CO), 147.5 (12-C), 147.2 (8-C), 128.2 (13-C), 109.6 (17-C), 79.9 (3-C), 73.3 (15-C), 67.5 (14-C), 64.2 (19-C), 56.2 (9-C), 55.3 (5-C), 42.8 (4-C), 39.1 (10-C), 38.4 (7-C), 37.6 (1-C), 26.1 (SiC(CH3)3), 25.8 (SiC(CH3)3), 25.6 (2-C), 24.9 (6-C), 24.8 (11-C), 23.4 (18-C), 20.9 (CH3CO), 18.5 (SiC(CH3)3), 18.0 (SiC(CH3)3), 15.0 (20-C), −4.3 (SiCH3), −4.7 (SiCH3), −5.47 (SiCH3), −5.53 (SiCH3); HRMS (ESI) m/z 643.3841 [M + Na]+, calculated for C34H60O6Si2Na, 643.3826.
3-Acetoxy-14β,19-di-tert-butyldimethylsilyloxy-andrographolide (9b). 61% yield, white solid, mp 157.8–159.6 °C. 1H NMR (400 MHz, C6D6) δ 7.00–6.94 (1H, m, 12-H), 4.86 (1H, s, 17α-H), 4.76 (1H, dd, J = 11.3, 5.2 Hz, 3β-H), 4.58–4.49 (1H, m, 14α-H), 4.39 (1H, s, 17β-H), 3.93 (1H, d, J = 10.5 Hz, 19α-H), 3.81–3.65 (3H, m, 15-H and 19β-H), 2.50–2.24 (3H, m, 9β-H and 11-H), 1.82–1.74 (3H, m, 2α-H and 7-H), 1.74 (3H, s, CH3CO), 1.72–1.60 (2H, m, 2β-H and 1α-H), 1.58–1.49 (2H, m, 1β-H and 6α-H), 1.04 (3H, s, 18-H), 1.02–0.99 (2H, m, 5β-H and 6β-H), 0.98 (9H, s, SiC(CH3)3), 0.90 (3H, s, 20-H), 0.87 (9H, s, SiC(CH3)3), 0.06 (6H, d, J = 4.6 Hz, Si(CH3)2), −0.00 (3H, s, SiCH3), −0.16 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 169.8 (16-C), 169.0 (CH3CO), 148.6 (12-C), 147.3 (8-C), 128.4 (13-C), 108.0 (17-C), 79.7 (3-C), 73.3 (15-C), 67.8 (14-C), 64.4 (19-C), 56.0 (9-C), 55.1 (5-C), 42.7 (4-C), 39.1 (10-C), 38.5 (7-C), 37.4 (1-C), 26.1 (SiC(CH3)3), 25.9 (2-C), 25.7 (SiC(CH3)3), 25.5 (6-C), 24.7 (11-C), 23.3 (18-C), 20.8 (CH3CO), 18.5 (SiC(CH3)3), 18.0 (SiC(CH3)3), 14.9 (20-C), −4.2 (SiCH3), −4.8 (SiCH3), −5.49 (SiCH3), −5.55 (SiCH3); HRMS (ESI) m/z 643.3822 [M + Na]+, calculated for C34H60O6Si2Na, 643.3826.
Preparation of the compounds 10a and 10b
Under N2 atmosphere, 5.0 g (8.6 mmol) of compound 8a or 8b and 2.5 ml (21.6 mmol) of 2,6-lutidine were dissolved in 60.0 ml anhydrous dichloromethane. The solution was cooled to 0 °C and then 4.0 ml (17.3 mmol) of TBSOTf was added dropwise over 2 min. The reaction mixture was stirred for 1 h at 0 °C and then treated with ethyl acetate and sat. NaHCO3 solution after the reaction was complete. The organic phase was washed with brine for 6 times, dried over anhydrous Na2SO4, filtered and then concentrated. The residue was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/30) to yield compound 10a or 10b.
3,14α,19-Tri-tert-butyldimethylsilyloxy-andrographolide (10a). 71% yield, white solid, mp 154.6–155.3 °C. 1H NMR (400 MHz, C6D6) δ 7.10–7.05 (1H, m, 12-H), 4.87 (1H, s, 17α-H), 4.68 (1H, s, 17β-H), 4.57–4.51 (1H, m, 14β-H), 4.07 (1H, d, J = 10.6 Hz, 19α-H), 3.82 (2H, dd, J = 9.7, 6.7 Hz, 15α-H and 19β-H), 3.72 (1H, dd, J = 9.6, 3.5 Hz, 15β-H), 3.29 (1H, dd, J = 11.6, 4.1 Hz, 3β-H), 2.67–2.55 (1H, m, 11α-H), 2.42–2.23 (2H, m, 9β-H and 11β-H), 1.99–1.53 (7H, m, 1-H, 2-H, 6α-H and 7-H), 1.16 (3H, s, 18-H), 1.05 (1H, d, J = 3.5 Hz, 5β-H), 1.03 (9H, s, SiC(CH3)3), 0.99 (1H, m, 6β-H), 0.97 (12H, d, J = 8.7 Hz, 20-H and SiC(CH3)3), 0.86 (9H, s, SiC(CH3)3), 0.15 (3H, s, SiCH3), 0.10 (6H, d, J = 2.6 Hz, Si(CH3)2), 0.07 (3H, s, SiCH3), 0.02 (3H, s, SiCH3), −0.15 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 169.0 (16-C), 147.9 (12-C), 147.5 (8-C), 128.4 (13-C), 109.3 (17-C), 79.7 (3-C), 73.3 (15-C), 67.6 (14-C), 64.8 (19-C), 56.6 (9-C), 55.3 (5-C), 44.2 (4-C), 39.2 (10-C), 38.9 (7-C), 38.0 (1-C), 28.8 (2-C), 26.5 (6-C), 26.19 (SiC(CH3)3), 26.15 (SiC(CH3)3), 25.8 (SiC(CH3)3), 24.9 (11-C), 24.0 (18-C), 18.5 (SiC(CH3)3), 18.4 (SiC(CH3)3), 18.0 (SiC(CH3)3), 14.9 (20-C), −3.7 (SiCH3), −4.3 (SiCH3), −4.7 (Si(CH3)2), −5.4 (SiCH3), −5.5 (SiCH3); HRMS (ESI) m/z 715.4593 [M + Na]+, calculated for C38H72O5Si3Na, 715.4585.
3,14β,19-Tri-tert-butyldimethylsilyl-oxy-andrographolide (10b). 89% yield, white solid, mp 159.2–160.5 °C. 1H NMR (400 MHz, C6D6) δ 7.01 (1H, ddd, J = 8.6, 4.4, 2.0 Hz, 12-H), 4.88 (1H, s, 17α-H), 4.61–4.54 (1H, m, 14α-H), 4.41 (1H, s, 17β-H), 4.06 (1H, d, J = 10.6 Hz, 19α-H), 3.81 (1H, d, J = 10.5 Hz, 19β-H), 3.77 (1H, dd, J = 9.6, 6.4 Hz, 15α-H), 3.71 (1H, dd, J = 9.6, 3.6 Hz, 15β-H), 3.24 (1H, dd, J = 11.7, 4.3 Hz, 3β-H), 2.56–2.34 (3H, m, 9β-H and 11-H), 2.06–1.74 (3H, m, 2α-H and 7-H), 1.73–1.46 (4H, m, 1-H, 2β-H and 6α-H), 1.15 (3H, s, 18-H), 1.02 (9H, s, SiC(CH3)3), 0.99 (9H, s, SiC(CH3)3), 0.97 (3H, s, 20-H), 0.93 (2H, dt, J = 10.2, 4.9 Hz, 5β-H and 6β-H), 0.88 (9H, s, SiC(CH3)3), 0.14 (3H, s, SiCH3), 0.12–0.06 (9H, m, SiCH3 and Si(CH3)2), 0.02 (3H, s, SiCH3), −0.15 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 169.1 (16-C), 149.0 (12-C), 147.6 (8-C), 128.4 (13-C), 107.7 (17-C), 79.6 (3-C), 73.4 (15-C), 67.8 (14-C), 64.9 (19-C), 56.6 (9-C), 55.1 (5-C), 44.1 (4-C), 39.3 (10-C), 38.9 (7-C), 37.8 (1-C), 28.7 (2-C), 26.7 (6-C), 26.2 (SiC(CH3)3), 26.1 (SiC(CH3)3), 25.8 (SiC(CH3)3), 25.6 (11-C), 23.9 (18-C), 18.5 (SiC(CH3)3), 18.3 (SiC(CH3)3), 18.0 (SiC(CH3)3), 14.9 (20-C), −3.7 (SiCH3), −4.2 (SiCH3), −4.7 (SiCH3), −4.8 (SiCH3), −5.4 (SiCH3), −5.5 (SiCH3); HRMS (ESI) m/z 715.4579 [M + Na]+, calculated for C38H72O5Si3Na, 715.4585.
Preparation of the compounds 11a and 11b
To the solution of 1.0 g (1.6 mmol) of compound 9a or 9b in 30.0 ml anhydrous dichloromethane at −20 °C, 10.0 ml (6.7 mmol) of TFA and 1.00 ml (6.7 mmol) of H2O were added dropwise over 10 min. The reaction mixture was stirred for 0.5 h at −20 °C, and then diluted with ethyl acetate and carefully treated with sat. NaHCO3 solution. The organic phase was washed with brine, dried over anhydrous Na2SO4, filtered and then concentrated. After silica gel column chromatography (ethyl acetate/petroleum ether 1/7), compound 11a or 11b was afforded.
3-Acetoxy-14α-tert-butyl-dimethylsilyloxy-andrographolide (11a). 64% yield, white solid, mp 113.9–115.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.65–6.59 (1H, m, 12-H), 5.20 (1H, d, J = 5.7 Hz, 14β-H), 4.81 (1H, s, 17α-H), 4.52 (1H, s, 17β-H), 4.52–4.44 (2H, m, 3β-H and 15α-H), 4.01 (1H, dd, J = 7.3, 2.7 Hz, 15β-H), 3.99 (1H, d, J = 5.3 Hz, 19-OH), 3.64 (1H, dd, J = 11.4, 6.1 Hz, 19α-H), 3.54 (1H, dd, J = 11.4, 4.7 Hz, 19β-H), 2.46 (2H, t, J = 8.0 Hz, 9β-H and 11α-H), 2.31 (1H, dt, J = 12.9, 3.2 Hz, 11β-H), 2.01 (3H, s, CH3CO), 2.01–1.98 (1H, m, 7α-H), 1.93 (1H, td, J = 13.3, 12.8, 4.5 Hz, 7β-H), 1.81–1.53 (5H, m, 1-H, 2-H and 6α-H), 1.40–1.28 (2H, m, 5β-H and 6β-H), 0.91 (3H, s, 18-H), 0.87 (9H, s, SiC(CH3)3), 0.75 (3H, s, 20-H), 0.15 (3H, s, SiCH3), 0.12 (3H, s, SiCH3); 13C NMR (101 MHz, DMSO-d6) δ 170.1 (16-C), 169.4 (CH3CO), 147.9 (12-C), 147.4 (8-C), 127.6 (13-C), 108.4 (17-C), 79.9 (3-C), 73.9 (15-C), 66.4 (14-C), 61.6 (19-C), 55.1 (9-C), 54.4 (5-C), 42.0 (4-C), 38.5 (10-C), 37.5 (7-C), 36.7 (1-C), 25.5 (SiC(CH3)3), 24.7 (2-C), 24.2 (6-C), 23.9 (11-C), 22.7 (18-C), 20.9 (CH3CO), 17.5 (SiC(CH3)3), 14.3 (20-C), −4.5 (SiCH3), −5.0 (SiCH3); HRMS (ESI) m/z 529.2979 [M + Na]+, calculated for C28H46O6SiNa, 529.2961.
3-Acetoxy-14β-tert-butyldimethyl-silyloxy-andrographolide (11b). 66% yield, white solid, mp 163.2–164.9 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.58 (1H, dd, J = 7.2, 3.9 Hz, 12-H), 5.24 (1H, d, J = 5.1 Hz, 14α-H), 4.79 (1H, s, 17α-H), 4.50–4.45 (2H, m, 3β-H and 15α-H), 4.32 (1H, s, 17β-H), 4.05 (1H, s, 19-OH), 3.99 (1H, dd, J = 9.9, 2.2 Hz, 15β-H), 3.63 (1H, dd, J = 11.3, 4.6 Hz, 19α-H), 3.57–3.48 (1H, m, 19β-H), 2.48–2.24 (3H, m, 9β-H and 11-H), 2.05 (1H, d, J = 10.0 Hz, 7α-H), 2.00 (3H, s, CH3CO), 1.92 (1H, td, J = 12.9, 4.5 Hz, 7β-H), 1.82–1.49 (5H, m, 1-H, 2-H and 6α-H), 1.35 (2H, t, J = 13.0 Hz, 5β-H and 6β-H), 0.91 (3H, s, 18-H), 0.86 (9H, s, SiC(CH3)3), 0.75 (3H, s, 20-H), 0.13 (6H, d, J = 17.8 Hz, Si(CH3)2); 13C NMR (101 MHz, DMSO-d6) δ 170.1 (16-C), 169.4 (CH3CO), 148.0 (12-C), 147.4 (8-C), 128.1 (13-C), 107.9 (17-C), 79.8 (3-C), 73.9 (15-C), 66.7 (14-C), 61.6 (19-C), 54.9 (9-C), 54.4 (5-C), 42.0 (4-C), 38.6 (10-C), 37.6 (7-C), 36.7 (1-C), 25.6 (SiC(CH3)3), 24.9 (2-C), 24.8 (6-C), 24.0 (11-C), 22.7 (18-C), 21.0 (CH3CO), 17.6 (SiC(CH3)3), 14.4 (20-C), −4.3 (SiCH3), −5.0 (SiCH3); HRMS (ESI) m/z 529.2958 [M + Na]+, calculated for C28H46O6SiNa, 529.2961.
Preparation of the compounds 12a and 12b
To the solution of 0.5 g (0.7 mmol) of compound 10a or 10b in 10.0 ml anhydrous dichloromethane at −20 °C, 0.1 ml (1.3 mmol) of TFA was added dropwise over 2 min. The reaction mixture was stirred for 5 min at −20 °C and treated with ethyl acetate and sat. NaHCO3 solution after the reaction was complete. The organic phase was washed with brine, dried over anhydrous Na2SO4, filtered and then concentrated in vacuo. The residue was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/15) to obtain compound 12a or 12b.
3,14α-Di-tert-butyldimethylsilyloxy-andrographolide (12a). 71% yield, white solid, mp 176.6–177.7 °C. 1H NMR (400 MHz, C6D6) δ 7.00 (1H, ddd, J = 7.3, 5.3, 2.0 Hz, 12-H), 4.82 (1H, s, 17α-H), 4.66 (1H, s, 17β-H), 4.55–4.47 (1H, m, 14β-H), 4.24 (1H, d, J = 10.9 Hz, 19α-H), 3.81 (1H, dd, J = 9.6, 6.6 Hz, 15α-H), 3.70 (1H, dd, J = 9.6, 3.6 Hz, 15β-H), 3.42–3.30 (2H, m, 3β-H and 19-OH), 3.24 (1H, d, J = 10.9 Hz, 19β-H), 2.52 (1H, ddd, J = 17.6, 11.3, 7.6 Hz, 11α-H), 2.23–2.11 (2H, m, 9β-H and 11β-H), 1.84–1.70 (2H, m, 7-H), 1.66–1.43 (4H, m, 1-H and 2-H), 1.24 (3H, s, 18-H), 1.08 (1H, qd, J = 13.1, 4.2 Hz, 6α-H), 0.95 (9H, s, SiC(CH3)3), 0.90 (1H, d, J = 2.4 Hz, 6β-H), 0.87 (9H, s, SiC(CH3)3), 0.83 (1H, d, J = 3.7 Hz, 5β-H), 0.58 (3H, s, 20-H), 0.10 (3H, s, SiCH3), 0.06 (3H, s, SiCH3), 0.00 (3H, s, SiCH3), −0.16 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 169.0 (16-C), 147.3 (12-C), 147.2 (8-C), 128.2 (13-C), 109.8 (17-C), 82.4 (3-C), 73.3 (15-C), 67.6 (14-C), 64.1 (19-C), 56.1 (9-C), 55.0 (5-C), 43.7 (4-C), 38.7 (10-C), 38.0 (7-C), 37.4 (1-C), 28.6 (2-C), 26.0 (SiC(CH3)3), 25.8 (SiC(CH3)3), 24.9 (6-C), 24.1 (11-C), 23.6 (18-C), 18.1 (SiC(CH3)3), 18.0 (SiC(CH3)3), 15.5 (20-C), −4.1 (SiCH3), −4.3 (SiCH3), −4.7 (SiCH3), −4.9 (SiCH3); HRMS (ESI) (m/z): 601.3733 [M + Na]+, calculated for C32H58O5Si2Na, 601.3720.
3,14β-Di-tert-butyldimethylsilyloxy-andrograph-olide (12b). 50% yield, white solid, mp 175.8–177.0 °C. 1H NMR (400 MHz, C6D6) δ 6.96 (1H, ddd, J = 8.4, 4.5, 1.8 Hz, 12-H), 4.83 (1H, s, 17α-H), 4.59–4.49 (1H, m, 14α-H), 4.37 (1H, s, 17β-H), 4.23 (1H, d, J = 11.0 Hz, 19α-H), 3.76 (1H, dd, J = 16.0, 1.3 Hz, 15α-H), 3.70 (1H, dd, J = 9.5, 3.4 Hz, 15β-H), 3.37 (1H, t, J = 11.1 Hz, 19-OH), 3.30 (1H, dd, J = 11.5, 4.2 Hz, 3β-H), 3.24 (1H, d, J = 10.2 Hz, 19β-H), 2.47–2.16 (3H, m, 9β-H and 11-H), 1.74 (2H, tdd, J = 17.2, 13.2, 4.2 Hz, 7-H), 1.65–1.37 (4H, m, 1-H and 2-H), 1.25 (3H, s, 18-H), 1.20–1.05 (1H, m, 6α-H), 0.94 (9H, s, SiC(CH3)3), 0.87 (9H, s, SiC(CH3)3), 0.86–0.74 (2H, m, 5β-H and 6β-H), 0.59 (3H, s, 20-H), 0.09 (3H, s, SiCH3), 0.04 (3H, s, SiCH3), −0.00 (3H, s, SiCH3), −0.16 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 169.0 (16-C), 148.2 (12-C), 147.2 (8-C), 128.5 (13-C), 108.1 (17-C), 82.4 (3-C), 73.4 (15-C), 67.8 (14-C), 64.1 (19-C), 55.9 (9-C), 54.9 (5-C), 43.7 (4-C), 38.7 (10-C), 38.0 (7-C), 37.3 (1-C), 28.5 (2-C), 26.0 (SiC(CH3)3), 25.8 (SiC(CH3)3), 25.6 (6-C), 24.1 (11-C), 23.6 (18-C), 18.1 (SiC(CH3)3), 18.0 (SiC(CH3)3), 15.5 (20-C), −4.1 (SiCH3), −4.2 (SiCH3), −4.8 (SiCH3), −4.9 (SiCH3); HRMS (ESI) m/z 579.3896 [M + H]+, calculated for C32H59O5Si2, 579.3901.
Preparation of the compounds 13a13 and 13b5
The preparation of compound 13a or 13b from 3a or 3b, respectively, used the same procedure that was used for the synthesis of 6a or 6b. Compound 13a or 13b was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/6).
3,19-Acetonylidene-14α-acetoxy-andrographolide (13a). 93% yield, white solid, mp 106.0–107.2 °C. 1H NMR (400 MHz, C6D6) δ 7.04 (1H, td, J = 6.9, 1.7 Hz, 12-H), 5.62 (1H, d, J = 6.0 Hz, 14β-H), 4.80 (1H, d, J = 1.1 Hz, 17α-H), 4.48 (1H, s, 17β-H), 3.90–3.81 (2H, m, 15α-H and 19α-H), 3.76 (1H, dd, J = 11.1, 2.0 Hz, 15β-H), 3.46 (1H, dd, J = 7.9, 3.5 Hz, 3β-H), 3.09 (1H, d, J = 11.5 Hz, 19β-H), 2.34–2.12 (3H, m, 9β-H and 11-H), 1.95–1.82 (1H, m, 7α-H), 1.72 (1H, td, J = 13.6, 13.1, 4.7 Hz, 7β-H), 1.65–1.58 (1H, m, 2α-H), 1.56 (3H, s, CH3CO), 1.55–1.52 (1H, m, 2β-H), 1.48 (1H, s, 1α-H), 1.45 (3H, s, CCH3), 1.40 (3H, s, CCH3), 1.38–1.31 (1H, m, 1β-H), 1.11 (3H, s, 18-H), 1.06–0.93 (2H, m, 5β-H and 6α-H), 0.90 (1H, dd, J = 12.9, 2.2 Hz, 6β-H), 0.86 (3H, s, 20-H); HRMS (ESI) m/z 455.2418 [M + Na]+, calculated for C25H36O6Na, 455.2410.
3,19-Acetonylidene-14β-acetoxy-andrographolide (13b). 90% yield, white solid, mp 140.0–142.0 °C. 1H NMR (400 MHz, CDCl3) δ 7.00 (1H, td, J = 6.9, 1.3 Hz, 12-H), 5.93 (1H, d, J = 6.0 Hz, 14α-H), 4.87 (1H, s, 17α-H), 4.56 (1H, dd, J = 11.3, 6.2 Hz, 15α-H), 4.41 (1H, s, 17β-H), 4.22 (1H, dd, J = 11.3, 1.8 Hz, 15β-H), 3.94 (1H, d, J = 11.6 Hz, 19α-H), 3.49 (1H, dd, J = 8.3, 3.8 Hz, 3β-H), 3.16 (1H, d, J = 11.6 Hz, 19β-H), 2.56–2.31 (3H, m, 9β-H and 11-H), 2.10 (3H, s, CH3CO), 2.03–1.92 (2H, m, 7-H), 1.88 (1H, d, J = 10.5 Hz, 2α-H), 1.83–1.64 (3H, m, 1-H and 2β-H), 1.39 (3H, s, CCH3), 1.35 (3H, s, CCH3), 1.32–1.22 (3H, m, 5β-H and 6-H), 1.18 (3H, s, 18-H), 0.94 (3H, s, 20-H); HRMS (ESI) m/z 455.2397 [M + Na]+, calculated for C25H36O6Na, 455.2410.
Preparation of the compounds 14a13 and 14b5
The procedure of the synthesis of 5a or 5b was used for the preparation of compound 14a or 14b from 13a or 13b. Crude product was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/2) to obtain compound 14a or 14b.
14α-Acetoxy-andrographolide (14a). 93% yield, white solid, mp 169.8–170.8 °C. 1H NMR (400 MHz, CD3OD) δ 6.98–6.91 (1H, m, 12-H), 6.02 (1H, d, J = 6.0 Hz, 14β-H), 4.89 (1H, s, 17α-H), 4.57 (1H, dd, J = 11.1, 6.1 Hz, 15α-H), 4.55 (1H, s, 17β-H), 4.29 (1H, dd, J = 11.1, 1.8 Hz, 15β-H), 4.11 (1H, d, J = 11.1 Hz, 19α-H), 3.41 (1H, d, J = 7.8 Hz, 3β-H), 3.37 (1H, d, J = 11.2 Hz, 19β-H), 2.57 (1H, ddd, J = 16.8, 6.3, 3.4 Hz, 11α-H), 2.49–2.39 (2H, m, 9β-H and 11β-H), 2.10 (3H, s, CH3CO), 2.08–1.92 (2H, m, 7-H), 1.90–1.74 (4H, m, 1-H and 2-H), 1.45–1.24 (3H, m, 5β-H and 6-H), 1.22 (3H, s, 18-H), 0.73 (3H, s, 20-H); HRMS (ESI) m/z 415.2108 [M + Na]+, calculated for C20H30O5Na, 415.2097.
14β-Acetoxy-andrographolide (14b). 88% yield, white solid, mp 163.0–165.0 °C. 1H NMR (400 MHz, CDCl3) δ 6.98 (1H, td, J = 7.1, 1.6 Hz, 12-H), 5.91 (1H, d, J = 6.0 Hz, 14α-H), 4.85 (1H, s, 17α-H), 4.54 (1H, dd, J = 11.3, 6.1 Hz, 15α-H), 4.36 (1H, s, 17β-H), 4.23 (1H, dd, J = 11.3, 1.8 Hz, 15β-H), 4.16 (1H, d, J = 10.9 Hz, 19α-H), 3.46 (1H, dt, J = 9.4, 4.1 Hz, 3β-H), 3.37–3.26 (1H, m, 3α-OH), 3.03–2.86 (2H, m, 19β-H and 19-OH), 2.55–2.25 (3H, m, 9β-H and 11-H), 2.11 (3H, s, CH3CO), 1.96 (1H, td, J = 12.9, 12.3, 4.6 Hz, 7α-H), 1.89–1.75 (4H, m, 1α-H, 2-H and 7β-H), 1.75–1.67 (1H, m, 1β-H), 1.35–1.25 (1H, m, 6α-H), 1.24 (3H, s, 18-H), 1.23–1.13 (2H, m, 5β-H and 6β-H), 0.66 (3H, s, 20-H); HRMS (ESI) m/z 415.2067, [M + Na]+, calculated for C22H32O6Na, 415.2097.
Preparation of the compounds 15a14 and 15b
4.0 g (10.2 mmol) of compound 14a or 14b and 8.49 ml (61.2 mmol) of TEA were dissolved in 20.0 ml anhydrous dichloromethane and then 8.47 g (56.1 mmol) of TBSCl in 5.0 ml of anhydrous dichloromethane was added dropwise over 2 min. The reaction mixture was stirred for 1 h at room temperature and treated with ethyl acetate and sat. NaHCO3 solution after the reaction was complete. The organic phase was washed with brine, dried over anhydrous Na2SO4, filtered and then concentrated under reduced pressure. Compound 15a or 15b was obtained after purification using silica gel column chromatography (ethyl acetate/petroleum ether 1/6).
14α-Acetoxy-19-tert-butyldimethylsilyloxy-andrographolide (15a). 74% yield, white solid, mp 159.1–159.7 °C. 1H NMR (400 MHz, C6D6) δ 7.01 (1H, td, J = 6.8, 1.7 Hz, 12-H), 5.62 (1H, d, J = 6.0 Hz, 14β-H), 4.78 (1H, s, 17α-H), 4.45 (1H, s, 17β-H), 4.17 (1H, d, J = 10.0 Hz, 19α-H), 3.91–3.83 (2H, m, 15α-H and 19-OH), 3.76 (1H, dd, J = 11.1, 1.9 Hz, 15β-H), 3.43–3.34 (2H, m, 3β-H and 19β-H), 2.24–2.15 (3H, m, 9β-H and 11-H), 1.97 (1H, dq, J = 13.6, 3.8 Hz, 7α-H), 1.79–1.65 (2H, m, 2α-H and 7β-H), 1.60 (1H, dt, J = 5.0, 2.3 Hz, 2β-H), 1.56 (3H, s, CH3CO), 1.49–1.42 (2H, m, 1-H), 1.29 (3H, s, 18-H), 1.11 (1H, qd, J = 12.9, 4.2 Hz, 6α-H), 0.98–0.93 (1H, m, 5β-H), 0.92 (9H, s, SiC(CH3)3), 0.88 (1H, d, J = 3.3 Hz, 6β-H), 0.55 (3H, s, 20-H), 0.00 (6H, d, J = 3.4 Hz, Si(CH3)2); HRMS (ESI) m/z 529.2972 [M + Na]+, calculated for C28H46O6SiNa, 529.2961.
14β-Acetoxy-19-tert-butyl-di-methylsilyloxy-andrographolide (15b). 69% yield, white solid, mp 147.9–149.8 °C. 1H NMR (400 MHz, C6D6) δ 7.03 (1H, t, J = 6.9 Hz, 12-H), 5.60 (1H, d, J = 5.2 Hz, 14α-H), 4.80 (1H, s, 17α-H), 4.38 (1H, s, 17β-H), 4.17 (1H, d, J = 10.0 Hz, 19α-H), 3.91 (1H, d, J = 6.7 Hz, 19-OH), 3.82 (1H, dd, J = 11.4, 6.1 Hz, 15α-H), 3.77 (1H, d, J = 10.9 Hz, 15β-H), 3.40 (1H, d, J = 10.5 Hz, 19β-H), 3.35 (1H, dd, J = 13.0, 5.5 Hz, 3β-H), 2.27–2.06 (3H, m, 9β-H and 11-H), 1.92 (1H, dd, J = 13.5, 3.8 Hz, 7α-H), 1.69 (2H, q, J = 14.7, 13.7 Hz, 2α-H and 7β-H), 1.57 (3H, s, CH3CO), 1.55 (1H, dd, J = 3.4, 2.7 Hz, 2β-H), 1.45–1.34 (2H, m, 1-H), 1.28 (3H, s, 18-H), 1.11 (1H, qd, J = 12.5, 12.0, 4.3 Hz, 6α-H), 0.91 (9H, s, SiC(CH3)3), 0.90–0.84 (2H, m, 5β-H and 6β-H), 0.56 (3H, s, 20-H), 0.01 (6H, d, J = 3.5 Hz, Si(CH3)2); 13C NMR (101 MHz, C6D6) δ 169.7 (16-C), 168.5 (CH3CO), 149.3 (12-C), 147.6 (8-C), 125.0 (13-C), 108.0 (17-C), 79.8 (3-C), 71.2 (15-C), 68.1 (14-C), 65.5 (19-C), 55.5 (9-C), 55.1 (5-C), 42.9 (4-C), 39.2 (10-C), 38.0 (7-C), 37.1 (1-C), 29.2 (2-C), 26.0 (SiC(CH3)3), 25.5 (6-C), 24.2 (11-C), 23.5 (18-C), 20.2 (CH3CO), 18.3 (SiC(CH3)3), 15.4 (20-C), −5.71 (SiCH3), −5.74 (SiCH3); HRMS (ESI) m/z 507.3139 [M + H]+, calculated for C28H47O6Si, 507.3142.
Preparation of the compounds 16a and 16b
3.5 g (3.9 mmol) of compound 15a or 15b and 3.35 g (13.8 mmol) of DMP were dissolved in 30.0 ml anhydrous dichloromethane. The reaction mixture, which was protected from light, was stirred for 1 h at room temperature and treated with ethyl acetate and sat. Na2S2O3 solution after the reaction was complete. The organic phase was washed with sat. NaHCO3 and brine, dried over anhydrous Na2SO4, filtered, concentrated, and then purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/7) to yield compound 16a or 16b.
3-Oxo-14α-acetoxy-19-tert-butyldi-methylsilyloxy-andrographolide (16a). 91% yield, white solid, mp 122.0–122.7 °C. 1H NMR (400 MHz, C6D6) δ 6.98 (1H, ddd, J = 7.6, 6.2, 1.7 Hz, 12-H), 5.63 (1H, d, J = 5.9 Hz, 14β-H), 4.80 (1H, s, 17α-H), 4.48 (1H, s, 17β-H), 3.87 (1H, dd, J = 11.1, 6.0 Hz, 15α-H), 3.79–3.71 (2H, m, 15β-H and 19α-H), 3.54 (1H, d, J = 9.9 Hz, 19β-H), 2.45 (1H, ddd, J = 15.1, 14.0, 5.9 Hz, 2α-H), 2.31 (1H, ddd, J = 15.2, 4.9, 3.0 Hz, 11α-H), 2.27–2.08 (3H, m, 2β-H, 9β-H and 11β-H), 1.72 (1H, td, J = 13.8, 12.2, 5.3 Hz, 7α-H), 1.56 (4H, m, CH3CO and 7β-H), 1.51–1.29 (4H, m, 1-H, 5β-H and 6α-H), 1.17 (3H, s, 18-H), 1.15–1.07 (1H, m, 6β-H), 0.93 (9H, s, SiC(CH3)3), 0.79 (3H, s, 20-H), 0.01 (6H, d, J = 1.3 Hz, Si(CH3)2); 13C NMR (101 MHz, C6D6) δ 211.2 (3-C), 169.8 (16-C), 168.4 (CH3CO), 149.1 (12-C), 147.3 (8-C), 124.9 (13-C), 109.2 (17-C), 71.1 (15-C), 67.8 (14-C), 66.7 (19-C), 56.3 (9-C), 55.1 (5-C), 53.7 (4-C), 38.8 (10-C), 37.9 (7-C), 37.5 (1-C), 36.3 (2-C), 26.0 (SiC(CH3)3), 25.6 (6-C), 25.2 (11-C), 22.2 (18-C), 20.1 (CH3CO), 18.5 (SiC(CH3)3), 14.4 (20-C), −5.5 (SiCH3), −5.6 (SiCH3); HRMS (ESI) m/z 527.2807 [M + Na]+, calculated for C28H44O6SiNa, 527.2805.
3-Oxo-14β-acetoxy-19-tert-butyldi-methylsilyloxy-andrographolide (16b). 86% yield, white solid, mp 123.2–124.5 °C. 1H NMR (400 MHz, C6D6) δ 7.00 (1H, td, J = 7.0, 1.6 Hz, 12-H), 5.62–5.55 (1H, m, 14α-H), 4.82 (1H, s, 17α-H), 4.40 (1H, s, 17β-H), 3.84–3.71 (3H, m, 15-H and 19α-H), 3.53 (1H, d, J = 9.9 Hz, 19β-H), 2.41 (1H, td, J = 14.6, 14.2, 5.9 Hz, 2α-H), 2.31–2.12 (4H, m, 2β-H, 9β-H and 11-H), 1.66 (1H, td, J = 12.3, 5.0 Hz, 7α-H), 1.55 (3H, s, CH3CO), 1.53–1.24 (5H, m, 1-H, 5β-H, 6α-H and 7β-H), 1.16 (3H, s, 18-H), 1.06 (1H, td, J = 13.5, 4.5 Hz, 6β-H), 0.94 (9H, s, SiC(CH3)3), 0.79 (3H, s, 20-H), 0.02 (6H, d, J = 1.3 Hz, Si(CH3)2); 13C NMR (101 MHz, C6D6) δ 211.2 (3-C), 169.7 (16-C), 168.5 (CH3CO), 149.0 (12-C), 147.3 (8-C), 125.1 (13-C), 108.7 (17-C), 71.2 (15-C), 68.1 (14-C), 66.6 (19-C), 56.2 (9-C), 54.7 (5-C), 53.7 (4-C), 39.0 (10-C), 37.8 (7-C), 37.4 (1-C), 36.2 (2-C), 26.0 (6-C), 25.8 (SiC(CH3)3), 25.1 (11-C), 22.2 (18-C), 20.2 (CH3CO), 18.5 (SiC(CH3)3), 14.4 (20-C), −5.5 (SiCH3), −5.6 (SiCH3); HRMS (ESI) m/z 505.2977 [M + H]+, calculated for C28H45O6Si, 505.2985.
Preparation of the compounds 17a and 17b
The procedure used for the preparation of compounds 12a or 12b was used for the preparation of compound 17a or 17b from 16a or 16b. Compound 17a or 17b was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/3).
3-Oxo-14α-acetoxy-andrographolide (17a). 93% yield, white solid, mp 142.0–142.9 °C. 1H NMR (400 MHz, CD3OD) δ 6.98–6.91 (1H, m, 12-H), 6.03 (1H, d, J = 6.0 Hz, 14β-H), 4.95 (1H, s, 17α-H), 4.62 (1H, s, 17β-H), 4.57 (1H, dd, J = 11.1, 6.1 Hz, 15α-H), 4.30 (1H, dd, J = 11.1, 1.8 Hz, 15β-H), 4.01 (1H, d, J = 11.2 Hz, 19α-H), 3.47 (1H, d, J = 11.2 Hz, 19β-H), 2.82 (1H, td, J = 14.5, 5.8 Hz, 2α-H), 2.61 (1H, ddd, J = 16.5, 6.3, 3.6 Hz, 11α-H), 2.57–2.43 (2H, m, 2β-H and 11β-H), 2.32 (1H, ddd, J = 14.7, 4.4, 3.2 Hz, 9β-H), 2.15–2.11 (1H, m, 7α-H), 2.11 (3H, s, CH3CO), 2.09–2.03 (2H, m, 1α-H and 7β-H), 1.82 (1H, ddt, J = 12.8, 5.1, 2.3 Hz, 1β-H), 1.75 (1H, dd, J = 12.7, 2.6 Hz, 6α-H), 1.69–1.51 (2H, m, 5β-H and 6β-H), 1.15 (3H, s, 18-H), 1.01 (3H, s, 20-H); 13C NMR (101 MHz, CD3OD) δ 216.8 (3-C), 172.0 (16-C), 171.4 (CH3CO), 151.0 (12-C), 148.6 (8-C), 125.9 (13-C), 109.6 (17-C), 73.2 (15-C), 69.3 (14-C), 65.9 (19-C), 58.0 (9-C), 56.5 (5-C), 55.6 (4-C), 40.1 (10-C), 39.3 (7-C), 38.7 (1-C), 36.8 (2-C), 26.5 (6-C), 25.8 (11-C), 20.8 (18-C), 20.6 (CH3CO), 15.1 (20-C); HRMS (ESI) m/z 413.1939 [M + Na]+, calculated for C22H30O6Na, 413.1940.
3-Oxo-14β-acetoxy-andrographolide (17b). 91% yield, white solid, mp 185.2–185.9 °C. 1H NMR (400 MHz, CD3OD) δ 6.96 (1H, td, J = 7.0, 1.7 Hz, 12-H), 6.04 (1H, d, J = 6.0 Hz, 14α-H), 4.95 (1H, d, J = 0.8 Hz, 17α-H), 4.63–4.53 (2H, m, 15α-H and 17β-H), 4.30 (1H, dd, J = 11.1, 1.8 Hz, 15β-H), 4.01 (1H, d, J = 11.2 Hz, 19α-H), 3.47 (1H, d, J = 11.2 Hz, 19β-H), 2.82 (1H, td, J = 14.5, 5.8 Hz, 2α-H), 2.67–2.41 (3H, m, 2β-H and 11-H), 2.31 (1H, ddd, J = 14.7, 4.4, 3.1 Hz, 9β-H), 2.14–2.10 (1H, m, 7α-H), 2.10 (3H, s, CH3CO), 2.08–2.02 (2H, m, 1α-H and 7β-H), 1.88–1.78 (1H, m, 1β-H), 1.74 (1H, dd, J = 12.7, 2.5 Hz, 6α-H), 1.67–1.51 (2H, m, 5β-H and 6β-H), 1.15 (3H, s, 18-H), 1.02 (3H, s, 20-H); 13C NMR (101 MHz, CD3OD) δ 216.8 (3-C), 171.9 (16-C), 171.4 (CH3CO), 151.1 (12-C), 148.5 (8-C), 126.1 (13-C), 109.4 (17-C), 73.2 (15-C), 69.6 (14-C), 65.9 (19-C), 58.2 (9-C), 56.5 (5-C), 55.7 (4-C), 40.3 (10-C), 39.4 (7-C), 38.7 (1-C), 36.7 (2-C), 26.8 (6-C), 25.9 (11-C), 20.8 (18-C), 20.7 (CH3CO), 15.1 (20-C); HRMS (ESI) m/z 413.1931 [M + Na]+, calculated for C22H30O6Na, 413.1940.
Preparation of the compounds 18a15 and 18b
The procedure used for the preparation of 6a or 6b was used for the preparation of compound 18a or 18b from 14a or 14b. Compound 18a or 18b was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/2).
14α,19-Diacetoxy-andrographolide (18a). 68% yield, white solid, mp 145.3–146.7 °C. 1H NMR (400 MHz, C6D6) δ 7.04–6.97 (1H, m, 12-H), 5.61 (1H, d, J = 5.7 Hz, 14β-H), 4.77 (1H, s, 17α-H), 4.44 (1H, s, 17β-H), 4.39 (1H, d, J = 11.8 Hz, 19α-H), 4.10 (1H, d, J = 11.7 Hz, 19β-H), 3.89–3.83 (1H, m, 15α-H), 3.75 (1H, d, J = 11.0 Hz, 15β-H), 3.15–3.05 (1H, m, 3β-H), 2.24–2.05 (3H, m, 9β-H and 11-H), 1.76–1.65 (2H, m, 7-H), 1.64 (3H, s, CH3CO), 1.60 (1H, dd, J = 8.4, 7.5 Hz, 2α-H), 1.56 (3H, s, CH3CO), 1.56–1.34 (4H, m, 1-H, 2β-H and 3α-OH), 1.24 (1H, qd, J = 13.0, 3.9 Hz, 6α-H), 1.11 (3H, s, 18-H), 0.90–0.77 (2H, m, 5β-H and 6β-H), 0.51 (3H, s, 20-H); HRMS (ESI) m/z 457.2215 [M + Na]+, calculated for C24H34O7Na, 457.2202.
14β,19-Diacetoxy-andro-grapholide (18b). 65% yield, white solid, mp 141.9–142.6 °C. 1H NMR (400 MHz, C6D6) δ 7.02 (1H, td, J = 7.0, 1.4 Hz, 12-H), 5.56 (1H, d, J = 2.8 Hz, 14α-H), 4.79 (1H, s, 17α-H), 4.40 (1H, d, J = 11.7 Hz, 19α-H), 4.36 (1H, s, 17β-H), 4.09 (1H, d, J = 11.8 Hz, 19β-H), 3.86–3.69 (2H, m, 15-H), 3.16–3.01 (1H, m, 3β-H), 2.22–2.05 (3H, m, 9β-H and 11-H), 1.83–1.64 (2H, m, 3α-OH and 7α-H), 1.62 (3H, s, CH3CO), 1.60 (1H, dd, J = 4.7, 3.0 Hz, 7β-H), 1.56 (3H, s, CH3CO), 1.55–1.41 (2H, m, 2-H), 1.39–1.17 (3H, m, 1-H and 6α-H), 1.11 (3H, s, 18-H), 0.78 (2H, dd, J = 31.6, 13.9 Hz, 5β-H and 6β-H), 0.52 (3H, s, 20-H); 13C NMR (101 MHz, C6D6) δ 170.5 (16-C), 169.8 (CH3CO), 168.7 (CH3CO), 149.4 (12-C), 147.5 (8-C), 124.9 (13-C), 108.1 (17-C), 78.5 (3-C), 71.3 (15-C), 68.2 (14-C), 65.0 (19-C), 55.5 (9-C), 55.0 (5-C), 42.6 (4-C), 39.3 (10-C), 38.0 (7-C), 37.1 (1-C), 28.2 (2-C), 25.5 (6-C), 24.6 (11-C), 22.8 (18-C), 20.6 (CH3CO), 20.2 (CH3CO), 14.6 (20-C); HRMS (ESI) m/z 457.2197 [M + Na]+, calculated for C24H34O7Na, 457.2202.
Preparation of the compounds 19a and 19b
The oxidation of compound 18a or 18b to 19a or 19b by DMP was used, which was similar to the procedure used for the preparation of 16a or 16b. Purification by silica gel column chromatography (ethyl acetate/petroleum ether 1/7) gave compound 19a or 19b.
3-Oxo-14α,19-diacetoxy-andro-grapholide (19a). 88% yield, white solid, mp 105.9–106.7 °C. 1H NMR (400 MHz, C6D6) δ 6.95–6.89 (1H, m, 12-H), 5.64–5.56 (1H, m, 14β-H), 4.76 (1H, s, 17α-H), 4.63 (1H, d, J = 11.2 Hz, 19α-H), 4.44 (1H, s, 17β-H), 3.86 (1H, dt, J = 11.1, 5.3 Hz, 15α-H), 3.75 (1H, dt, J = 11.1, 2.0 Hz, 15β-H), 3.70 (1H, d, J = 11.3 Hz, 19β-H), 2.64 (1H, td, J = 14.6, 5.8 Hz, 2α-H), 2.26 (1H, ddd, J = 14.8, 4.3, 3.0 Hz, 11α-H), 2.22–1.98 (3H, m, 2β-H, 9β-H and 11β-H), 1.63 (3H, s, CH3CO), 1.61 (1H, d, J = 4.8 Hz, 7α-H), 1.56 (3H, s, CH3CO), 1.53–1.44 (1H, m, 7β-H), 1.40–1.29 (2H, m, 1-H), 1.28–1.20 (1H, m, 6α-H), 1.20 (3H, s, 18-H), 1.12–0.98 (2H, m, 5β-H and 6β-H), 0.65 (3H, s, 20-H); 13C NMR (101 MHz, C6D6) δ 210.2 (3-C), 170.2 (16-C), 169.8 (CH3CO), 168.4 (CH3CO), 148.8 (12-C), 146.7 (8-C), 125.0 (13-C), 109.4 (17-C), 71.1 (15-C), 67.8 (14-C), 66.1 (19-C), 56.6 (9-C), 54.9 (5-C), 52.2 (4-C), 38.9 (10-C), 37.9 (7-C), 37.6 (1-C), 35.5 (2-C), 25.4 (6-C), 24.7 (11-C), 20.9 (18-C), 20.3 (CH3CO), 20.1 (CH3CO), 14.4 (20-C); HRMS (ESI) m/z 455.2046 [M + Na]+, calculated for C24H32O7Na, 455.2046.
3-Oxo-14β,19-diacetoxy-andrographolide (19b). 90% yield, white solid, mp 135.1–135.8 °C. 1H NMR (400 MHz, C6D6) δ 6.95 (1H, td, J = 7.0, 1.6 Hz, 12-H), 5.56 (1H, d, J = 5.5 Hz, 14α-H), 4.78 (1H, d, J = 0.6 Hz, 17α-H), 4.64 (1H, d, J = 11.3 Hz, 19α-H), 4.37 (1H, s, 17β-H), 3.82 (1H, dd, J = 11.1, 5.6 Hz, 15α-H), 3.77 (1H, dd, J = 11.1, 2.1 Hz, 15β-H), 3.70 (1H, d, J = 11.3 Hz, 19β-H), 2.63 (1H, td, J = 14.6, 5.8 Hz, 2α-H), 2.21 (1H, ddd, J = 7.4, 5.8, 3.6 Hz, 11α-H), 2.14–2.05 (3H, m, 2β-H, 9β-H and 11β-H), 1.63 (3H, s, CH3CO), 1.59 (1H, dd, J = 13.2, 5.5 Hz, 7α-H), 1.56 (3H, s, CH3CO), 1.45 (1H, ddd, J = 13.0, 5.8, 3.1 Hz, 7β-H), 1.33 (2H, dd, J = 9.0, 4.0 Hz, 1-H), 1.23–1.16 (4H, m, 6α-H and 18-H), 1.12–0.96 (2H, m, 5β-H and 6β-H), 0.68 (3H, s, 20-H); 13C NMR (101 MHz, C6D6) δ 210.4 (3-C), 170.3 (16-C), 169.8 (CH3CO), 168.5 (CH3CO), 148.8 (12-C), 146.7 (8-C), 125.2 (13-C), 108.8 (17-C), 71.2 (15-C), 68.1 (14-C), 66.1 (19-C), 56.5 (9-C), 54.6 (5-C), 52.2 (4-C), 39.1 (10-C), 37.9 (7-C), 37.5 (1-C), 35.4 (2-C), 25.6 (6-C), 24.7 (11-C), 20.8 (18-C), 20.3 (CH3CO), 20.2 (CH3CO), 14.5 (20-C); HRMS (ESI) m/z 455.2039 [M + Na]+, calculated for C24H32O7Na, 455.2046.
Preparation of the compounds 20a16 and 20b
5.0 g (11.6 mmol) of compound 19a or 19b was dissolved in 20.0 ml of methanol and then treated with 4.4 g (23.15 mmol) of p-TSA at 40 °C for 8 h. The mixture was diluted by ethyl acetate and washed with sat. NaHCO3 solution and brine; the organic phase was dried over anhydrous Na2SO4, filtered, and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/1) to afford compound 20a or 20b.
3-Oxo-andrographolide (20a). 51% yield, white solid, mp 193.4–194.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.63 (1H, td, J = 6.6, 1.5 Hz, 12-H), 5.75 (1H, d, J = 6.0 Hz, 14α-OH), 4.93 (1H, t, J = 5.7 Hz, 14β-H), 4.87 (1H, s, 17α-H), 4.70 (1H, s, 17β-H), 4.55 (1H, t, J = 5.4 Hz, 19-OH), 4.39 (1H, dd, J = 9.9, 6.1 Hz, 15α-H), 4.04 (1H, dd, J = 9.9, 2.0 Hz, 15β-H), 3.85 (1H, dd, J = 10.9, 5.7 Hz, 19α-H), 3.30 (1H, dd, J = 10.9, 5.2 Hz, 19β-H), 2.75 (1H, td, J = 14.4, 5.5 Hz, 2α-H), 2.53 (2H, t, J = 7.1 Hz, 2β-H and 11α-H), 2.40–2.30 (1H, m, 11β-H), 2.14 (1H, dt, J = 14.1, 3.4 Hz, 9β-H), 2.07–1.92 (3H, m, 1α-H and 7-H), 1.76–1.64 (1H, m, 1β-H), 1.64–1.40 (3H, m, 5β-H and 6-H), 1.01 (3H, s, 18-H), 0.93 (3H, s, 20-H); HRMS (ESI) m/z: 371.1846 [M + Na]+, calculated for C20H28O5Na, 371.1834.
3-Oxo-14β-andrographolide (20b). 53% yield, white solid, mp 152.4–153.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.67–6.57 (1H, m, 12-H), 5.68 (1H, d, J = 5.9 Hz, 14β-OH), 4.96 (1H, t, J = 5.5 Hz, 14α-H), 4.86 (1H, s, 17α-H), 4.56 (1H, t, J = 5.4 Hz, 19-OH), 4.49 (1H, s, 17β-H), 4.41 (1H, dd, J = 9.9, 6.2 Hz, 15α-H), 4.02 (1H, dd, J = 9.9, 2.2 Hz, 15β-H), 3.85 (1H, dd, J = 10.9, 5.7 Hz, 19α-H), 3.30 (1H, dd, J = 10.9, 5.2 Hz, 19β-H), 2.75 (1H, td, J = 14.4, 5.6 Hz, 2α-H), 2.60 (1H, ddd, J = 16.1, 5.8, 2.7 Hz, 11α-H), 2.48–2.30 (2H, m, 2β-H and 11β-H), 2.14 (1H, dt, J = 14.0, 3.5 Hz, 9β-H), 2.09–1.90 (3H, m, 1α-H and 7-H), 1.77–1.65 (1H, m, 1β-H), 1.64–1.40 (3H, m, 5β-H and 6-H), 1.01 (3H, s, 18-H), 0.94 (3H, s, 20-H); 13C NMR (101 MHz, DMSO-d6) δ 213.5 (3-C), 169.9 (16-C), 147.4 (12-C), 146.2 (8-C), 129.3 (13-C), 108.5 (17-C), 74.3 (15-C), 64.9 (14-C), 64.0 (19-C), 56.1 (9-C), 54.7 (5-C), 54.1 (4-C), 38.9 (10-C), 37.8 (7-C), 37.2 (1-C), 35.6 (2-C), 24.7 (6-C), 24.4 (11-C), 20.3 (18-C), 14.6 (20-C); HRMS (ESI) m/z: 371.1829 [M + Na]+, calculated for C20H28O5Na, 371.1834.
Preparation of the compounds 21a and 21b
The 19-silylation of 20a or 20b to 21a or 21b by TBSCl and imidazole at room temperature was conducted according to the procedure used for the preparation of 8a or 8b. Compound 21a or 21b was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/3).
3-Oxo-19-tert-butyl-dimethylsilyloxy-andrographolide (21a). 57% yield, white solid, mp 174.0–175.8 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.68–6.60 (1H, m, 12-H), 5.72 (1H, d, J = 5.2 Hz, 14α-OH), 4.93 (1H, s, 14β-H), 4.89 (1H, s, 17α-H), 4.71 (1H, s, 17β-H), 4.39 (1H, dd, J = 9.9, 6.1 Hz, 15α-H), 4.04 (1H, dd, J = 9.9, 2.0 Hz, 15β-H), 3.85 (1H, d, J = 10.1 Hz, 19α-H), 3.50 (1H, d, J = 10.1 Hz, 19β-H), 2.69–2.52 (3H, m, 2α-H and 11-H), 2.41–2.32 (1H, m, 9β-H), 2.27–2.18 (1H, m, 2β-H), 2.06–1.94 (3H, m, 1α-H and 7-H), 1.78–1.64 (2H, m, 1β-H and 6α-H), 1.63–1.43 (2H, m, 5β-H and 6β-H), 1.01 (3H, s, 18-H), 0.92 (3H, s, 20-H), 0.81 (9H, s, SiC(CH3)3), −0.02 (6H, s, Si(CH3)2); 13C NMR (101 MHz, DMSO-d6) δ 212.6 (3-C), 169.9 (16-C), 147.1 (12-C), 146.0 (8-C), 129.1 (13-C), 109.1 (17-C), 74.3 (15-C), 65.6 (14-C), 64.5 (19-C), 55.4 (9-C), 54.5 (5-C), 53.3 (4-C), 38.5 (10-C), 37.10 (7-C), 37.07 (1-C), 35.5 (2-C), 25.6 (SiC(CH3)3), 24.5 (6-C), 24.2 (11-C), 21.0 (18-C), 17.8 (SiC(CH3)3), 14.3 (20-C), −5.7 (Si(CH3)2); HRMS (ESI) m/z 485.2711 [M + Na]+, calculated for C26H42O5SiNa, 485.2699.
3-Oxo-19-tert-butyl-dimethyl-silyloxy-14β-andrographolide (21b). 55% yield, white solid, mp 126.1–127.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.67–6.59 (1H, m, 12-H), 5.68 (1H, d, J = 4.9 Hz, 14β-OH), 4.96 (1H, s, 14α-H), 4.87 (1H, s, 17α-H), 4.50 (1H, s, 17β-H), 4.41 (1H, dd, J = 9.9, 6.2 Hz, 15α-H), 4.02 (1H, dd, J = 9.9, 2.2 Hz, 15β-H), 3.85 (1H, d, J = 10.1 Hz, 19α-H), 3.49 (1H, d, J = 10.1 Hz, 19β-H), 2.69–2.55 (2H, m, 2α-H and 11α-H), 2.47–2.32 (2H, m, 2β-H and 11β-H), 2.28–2.17 (1H, m, 9β-H), 2.10–1.93 (3H, m, 1α-H and 7-H), 1.81–1.42 (4H, m, 1β-H, 5β-H and 6-H), 1.01 (3H, s, 18-H), 0.92 (3H, s, 20-H), 0.80 (9H, s, SiC(CH3)3), −0.03 (6H, s, Si(CH3)2); 13C NMR (101 MHz, DMSO-d6) δ 212.7 (3-C), 169.9 (16-C), 147.4 (12-C), 146.1 (8-C), 129.3 (13-C), 108.5 (17-C), 74.3 (15-C), 65.6 (14-C), 64.9 (19-C), 55.6 (9-C), 54.5 (5-C), 53.4 (4-C), 38.7 (10-C), 37.13 (7-C), 37.12 (1-C), 35.5 (2-C), 25.7 (SiC(CH3)3), 24.7 (6-C), 24.5 (11-C), 21.0 (18-C), 17.9 (SiC(CH3)3), 14.4 (20-C), −5.7 (Si(CH3)2); HRMS (ESI) m/z 485.2672 [M + Na]+, calculated for C26H42O5SiNa, 485.2699.
Preparation of the compounds 22a and 22b
The oxidation of compound 6a or 6b to 22a or 22b by DMP was used similar to the procedure used for the synthesis of 16a or 16b. Purification by silica gel column chromatography (ethyl acetate/petroleum ether 1/7) gave compound 22a or 22b.
3-Oxo-14α-tert-butyldimethylsilyloxy-19-acetoxy-andrographolide (22a). 88% yield, white solid, mp 98.3–99.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.64–6.57 (1H, m, 12-H), 5.22 (1H, d, J = 5.7 Hz, 14β-H), 4.88 (1H, s, 17α-H), 4.59 (1H, s, 17β-H), 4.53–4.43 (2H, m, 15α-H and 19α-H), 4.01 (1H, dd, J = 10.0, 2.3 Hz, 15β-H), 3.93 (1H, d, J = 11.3 Hz, 19β-H), 2.81 (1H, td, J = 14.6, 6.1 Hz, 2α-H), 2.62–2.51 (1H, m, 11α-H), 2.49–2.43 (1H, m, 11β-H), 2.41–2.34 (1H, m, 2β-H), 2.24 (1H, ddd, J = 14.8, 4.3, 2.8 Hz, 9β-H), 2.11 (1H, dd, J = 10.5, 3.3 Hz, 7α-H), 2.08–1.94 (2H, m, 1α-H and 7β-H), 1.92 (3H, s, CH3CO), 1.77 (2H, d, J = 10.4 Hz, 6α-H and 7β-H), 1.62 (1H, td, J = 13.5, 4.7 Hz, 6β-H), 1.46 (1H, qd, J = 13.6, 4.6 Hz, 5β-H), 1.06 (3H, s, 18-H), 0.91 (3H, s, 20-H), 0.88 (9H, s, SiC(CH3)3), 0.17 (3H, s, SiCH3), 0.13 (3H, s, SiCH3); 13C NMR (101 MHz, DMSO-d6) δ 211.9 (3-C), 170.1 (16-C), 169.4 (CH3CO), 147.0 (12-C and 8-C), 127.7 (13-C), 109.4 (17-C), 73.9 (15-C), 66.4 (14-C), 65.5 (19-C), 55.4 (9-C), 54.1 (5-C), 51.4 (4-C), 38.4 (10-C), 37.3 (7-C), 36.7 (1-C), 34.9 (2-C), 25.6 (SiC(CH3)3), 24.4 (6-C), 24.2 (11-C), 20.5 (18-C), 20.4 (CH3CO), 17.5 (SiC(CH3)3), 14.3 (20-C), −4.5 (SiCH3), −5.0 (SiCH3); HRMS (ESI) m/z: 527.2811 [M + Na]+, calculated for C28H44O6SiNa, 527.2805.
3-Oxo-14β-tert-butyldimethylsilyloxy-19-acetoxy-andrograph-olide (22b). 88% yield, white solid, mp 108.3–109.9 °C. 1H NMR (400 MHz, C6D6) δ 6.87–6.80 (1H, m, 12-H), 4.83 (1H, s, 17α-H), 4.68 (1H, d, J = 11.3 Hz, 19α-H), 4.54–4.47 (1H, m, 14α-H), 4.38 (1H, s, 17β-H), 3.76 (1H, dd, J = 9.6, 6.6 Hz, 15α-H), 3.73–3.66 (2H, m, 15β-H and 19β-H), 2.66 (1H, td, J = 14.6, 5.9 Hz, 2α-H), 2.28–2.19 (3H, m, 2β-H and 11-H), 2.18–2.08 (1H, m, 9β-H), 1.67 (1H, m, 7α-H), 1.63 (3H, s, CH3CO), 1.57 (1H, ddd, J = 14.5, 7.2, 4.2 Hz, 7β-H), 1.43–1.32 (2H, m, 1-H), 1.24 (1H, s, 6α-H), 1.21 (3H, s, 18-H), 1.16–0.99 (2H, m, 5β-H and 6β-H), 0.86 (9H, s, SiC(CH3)3), 0.78 (3H, s, 20-H), −0.03 (3H, s, SiCH3), −0.17 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 210.2 (3-C), 170.2 (16-C), 168.9 (CH3CO), 147.3 (12-C), 146.6 (8-C), 128.6 (13-C), 108.8 (17-C), 73.3 (15-C), 67.7 (14-C), 66.2 (19-C), 56.2 (9-C), 55.0 (5-C), 52.1 (4-C), 38.8 (10-C), 38.1 (7-C), 37.5 (1-C), 35.3 (2-C), 25.7 (SiC(CH3)3), 25.4 (6-C), 24.7 (11-C), 20.9 (18-C), 20.3 (CH3CO), 18.0 (SiC(CH3)3), 14.8 (20-C), −4.2 (SiCH3), −4.8 (SiCH3); HRMS (ESI) m/z: 527.2796 [M + Na]+, calculated for C28H44O6SiNa, 527.2805.
Preparation of the compounds 23a and 23b
1.0 g (2.0 mmol) of compound 22a or 22b was dissolved in 10.0 ml of tetrahydrofuran and then treated with 0.52 g (2.0 mmol) of TBAF at 0 °C for 3 h. The reaction mixture was treated with ethyl acetate and sat. NaHCO3 solution quickly after the reaction was complete. The organic phase was washed with brine, dried over anhydrous Na2SO4, filtered and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/3) to yield compound 23a or 23b.
3-Oxo-19-acetoxy-andrographolide (23a). 92% yield, white solid, mp 126.1–127.8 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.63 (1H, td, J = 6.6, 1.4 Hz, 12-H), 5.74 (1H, s, 14α-OH), 4.93 (1H, d, J = 5.6 Hz, 14β-H), 4.90 (1H, s, 17α-H), 4.72 (1H, s, 17β-H), 4.51 (1H, d, J = 11.3 Hz, 19α-H), 4.40 (1H, dd, J = 9.9, 6.1 Hz, 15α-H), 4.04 (1H, dd, J = 9.9, 2.1 Hz, 15β-H), 3.92 (1H, d, J = 11.3 Hz, 19β-H), 2.81 (1H, td, J = 14.6, 6.0 Hz, 2α-H), 2.55 (2H, t, J = 7.0 Hz, 2β-H and 11α-H), 2.38 (1H, dt, J = 10.5, 2.4 Hz, 11β-H), 2.24 (1H, ddd, J = 14.9, 4.5, 2.9 Hz, 9β-H), 2.09–1.96 (3H, m, 1α-H and 7-H), 1.92 (3H, s, CH3CO), 1.81–1.72 (2H, m, 1-H and 6α-H), 1.60 (1H, td, J = 13.5, 4.7 Hz, 5β-H), 1.46 (1H, qd, J = 13.2, 4.2 Hz, 6β-H), 1.06 (3H, s, 18-H), 0.92 (3H, s, 20-H); 13C NMR (101 MHz, DMSO-d6) δ 211.9 (3-C), 170.1 (16-C), 169.9 (CH3CO), 146.7 (12-C), 145.8 (8-C), 129.1 (13-C), 109.2 (17-C), 74.3 (15-C), 65.5 (14-C), 64.5 (19-C), 55.5 (9-C), 54.3 (5-C), 51.4 (4-C), 38.5 (10-C), 37.1 (7-C), 36.9 (1-C), 34.9 (2-C), 24.3 (6-C), 24.1 (11-C), 20.5 (18-C), 20.4 (CH3CO), 14.1 (20-C); HRMS (ESI) m/z 413.1951 [M + Na]+, calculated for C22H30O6Na, 413.1940.
3-Oxo-19-acetoxy-14β-andrographolide (23b). 87% yield, white solid, mp 144.3–145.6 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.66–6.59 (1H, m, 12-H), 5.69 (1H, d, J = 6.1 Hz, 14β-OH), 4.97 (1H, t, J = 6.0 Hz, 14α-H), 4.88 (1H, s, 17α-H), 4.57–4.47 (2H, m, 17β-H and 19α-H), 4.42 (1H, dd, J = 9.9, 6.2 Hz, 15α-H), 4.02 (1H, dd, J = 9.7, 2.4 Hz, 15β-H), 3.91 (1H, d, J = 11.3 Hz, 19β-H), 2.81 (1H, td, J = 14.5, 6.0 Hz, 2α-H), 2.62 (1H, ddd, J = 16.2, 5.6, 2.4 Hz, 11α-H), 2.49–2.31 (2H, m, 2β-H and 11β-H), 2.23 (1H, ddd, J = 14.7, 4.3, 2.8 Hz, 9β-H), 2.12–1.97 (3H, m, 1α-H and 7-H), 1.92 (3H, s, CH3CO), 1.81–1.69 (2H, m, 1β-H and 6α-H), 1.62 (1H, td, J = 13.5, 4.6 Hz, 5β-H), 1.53–1.37 (1H, m, 6β-H), 1.06 (3H, s, 18-H), 0.92 (3H, s, 20-H); 13C NMR (101 MHz, DMSO-d6) δ 212.1 (3-C), 170.2 (16-C), 169.9 (CH3CO), 147.0 (12-C), 146.1 (8-C), 129.4 (13-C), 108.7 (17-C), 74.3 (15-C), 65.6 (14-C), 64.9 (19-C), 55.7 (9-C), 54.3 (5-C), 51.5 (4-C), 38.8 (10-C), 37.2 (7-C), 36.9 (1-C), 34.9 (2-C), 24.7 (6-C), 24.3 (11-C), 20.6 (18-C), 20.4 (CH3CO), 14.2 (20-C); HRMS (ESI) m/z 413.1935 [M + Na]+, calculated for C22H30O6Na, 413.1940.
Preparation of the compounds 24a and 24b
The procedure used for the synthesis of 16a or 16b was used for the oxidation of 8a or 8b to 24a or 24b by DMP. The purification of compound 24a or 24b was conducted by silica gel column chromatography (ethyl acetate/petroleum ether 1/8).
3-Oxo-14α,19-di-tert-butyldimethylsilyloxy-andrographolide (24a). 91% yield, white solid, mp 94.6–95.2 °C. 1H NMR (400 MHz, C6D6) δ 6.94 (1H, ddd, J = 7.3, 5.2, 2.0 Hz, 12-H), 4.86 (1H, s, 17α-H), 4.71 (1H, s, 17β-H), 4.53–4.46 (1H, m, 14β-H), 3.88–3.78 (2H, m, 15α-H and 19α-H), 3.71 (1H, ddd, J = 9.7, 3.5, 1.6 Hz, 15β-H), 3.55 (1H, d, J = 9.9 Hz, 19β-H), 2.60–2.47 (2H, m, 2α-H and 11α-H), 2.40–2.31 (1H, m, 2β-H), 2.25–2.06 (2H, m, 9β-H and 11β-H), 1.82–1.62 (2H, m, 7-H), 1.57–1.42 (2H, m, 1-H), 1.41–1.30 (2H, m, 5β-H and 6α-H), 1.26–1.14 (4H, m, 6β-H and 18-H), 0.93 (9H, s, SiC(CH3)3), 0.91 (3H, s, 20-H), 0.87 (9H, s, SiC(CH3)3), 0.02 (6H, d, J = 1.6 Hz, Si(CH3)2), −0.01 (3H, s, SiCH3), −0.16 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 211.1 (3-C), 168.9 (16-C), 146.94 (12-C), 146.87 (8-C), 128.2 (13-C), 110.4 (17-C), 73.3 (15-C), 67.5 (14-C), 66.6 (19-C), 56.3 (9-C), 55.3 (5-C), 53.9 (4-C), 38.8 (10-C), 38.0 (7-C), 37.8 (1-C), 36.2 (2-C), 26.0 (SiC(CH3)3), 25.8 (SiC(CH3)3), 25.1 (6-C), 24.9 (11-C), 22.1 (18-C), 18.5 (SiC(CH3)3), 17.9 (SiC(CH3)3), 15.0 (20-C), −4.3 (SiCH3), −4.7 (SiCH3), −5.47 (SiCH3), −5.53 (SiCH3); HRMS (ESI) m/z 599.3577 [M + Na]+, calculated for C32H56O5Si2Na, 599.3564.
3-Oxo-14β,19-di-tert-butyl dimethyl-silyloxy-andrographolide (24b). 90% yield, white solid, mp 190.3–191.2 °C. 1H NMR (400 MHz, C6D6) δ 6.90 (1H, ddd, J = 7.1, 4.9, 2.0 Hz, 12-H), 4.87 (1H, d, J = 1.1 Hz, 17α-H), 4.57–4.50 (1H, m, 14α-H), 4.42 (1H, d, J = 0.9 Hz, 17β-H), 3.76 (2H, dd, J = 9.8, 6.7 Hz, 15α-H and 19α-H), 3.70 (1H, dd, J = 9.6, 3.6 Hz, 15β-H), 3.58 (1H, d, J = 9.9 Hz, 19β-H), 2.55–2.43 (1H, m, 2α-H), 2.43–2.20 (4H, m, 2β-H, 9β-H and 11-H), 1.80–1.60 (2H, m, 7-H), 1.56–1.28 (4H, m, 1-H, 5β-H and 6α-H), 1.22–1.07 (4H, m, 6β-H and 18-H), 0.93 (12H, s, 20-H and SiC(CH3)3), 0.86 (9H, s, SiC(CH3)3), 0.02 (6H, d, J = 1.3 Hz, Si(CH3)2), −0.01 (3H, s, SiCH3), −0.17 (3H, s, SiCH3); 13C NMR (101 MHz, C6D6) δ 211.1 (3-C), 169.0 (16-C), 148.0 (12-C), 146.9 (8-C), 128.5 (13-C), 108.7 (17-C), 73.3 (15-C), 67.7 (14-C), 66.6 (19-C), 55.9 (9-C), 55.1 (5-C), 53.7 (4-C), 38.8 (10-C), 37.9 (7-C), 37.7 (1-C), 36.1 (2-C), 26.0 (SiC(CH3)3), 25.7 (SiC(CH3)3), 25.6 (6-C), 25.2 (11-C), 22.2 (18-C), 18.5 (SiC(CH3)3), 18.0 (SiC(CH3)3), 14.8 (20-C), −4.2 (SiCH3), −4.8 (SiCH3), −5.46 (SiCH3), −5.54 (SiCH3); HRMS (ESI) m/z 599.3559 [M + Na]+, calculated for C32H56O5Si2Na, 599.3564.
Preparation of the compounds 25a and 25b
The deprotection of 19-OTBS of 24a or 24b was carried out using the procedure used for the synthesis of 12a or 12b for 2 h. Compound 25a or 25b was obtained after purification by silica gel column chromatography (ethyl acetate/petroleum ether 1/7).
3-Oxo-14α-tert-butyldimethyl-silyloxy-andrographolide (25a). 67% yield, white solid, mp 138.1–139.2 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.66–6.57 (1H, m, 12-H), 5.21 (1H, d, J = 5.6 Hz, 14β-H), 4.85 (1H, s, 17α-H), 4.57 (1H, s, 17β-H), 4.53 (1H, t, J = 4.6 Hz, 19-OH), 4.47 (1H, dd, J = 10.0, 5.9 Hz, 15α-H), 4.01 (1H, dd, J = 10.0, 2.3 Hz, 15β-H), 3.83 (1H, dd, J = 10.8, 4.7 Hz, 19α-H), 3.35 (1H, dd, J = 10.8, 4.7 Hz, 19β-H), 2.74 (1H, td, J = 14.4, 5.5 Hz, 2α-H), 2.59–2.51 (1H, m, 11α-H), 2.49–2.42 (1H, m, 2β-H), 2.40–2.31 (1H, m, 11β-H), 2.22–1.90 (4H, m, 1α-H, 7-H and 9β-H), 1.76–1.40 (4H, m, 1β-H, 5β-H and 6-H), 1.01 (3H, s, 18-H), 0.93 (3H, s, 20-H), 0.87 (9H, s, SiC(CH3)3), 0.17 (3H, s, SiCH3), 0.13 (3H, s, SiCH3); 13C NMR (101 MHz, DMSO-d6) δ 213.2 (3-C), 169.4 (16-C), 147.3 (12-C), 147.1 (8-C), 127.7 (13-C), 109.2 (17-C), 73.9 (15-C), 66.4 (14-C), 64.0 (19-C), 55.8 (9-C), 54.5 (5-C), 54.0 (4-C), 38.4 (10-C), 37.9 (7-C), 37.0 (1-C), 35.5 (2-C), 25.5 (SiC(CH3)3), 24.4 (6-C), 24.2 (11-C), 20.3 (18-C), 17.4 (SiC(CH3)3), 14.6 (20-C), −4.5 (SiCH3), −5.0 (SiCH3); HRMS (ESI) m/z 485.2710 [M + Na]+, calculated for C26H42O5SiNa, 485.2699.
3-Oxo-14β-tert-butyldimethylsilyl-oxy-andrograph-olide (25b). 75% yield, white solid, mp 152.1–152.8 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.58 (1H, dd, J = 6.6, 4.6 Hz, 12-H), 5.25 (1H, d, J = 5.7 Hz, 14α-H), 4.84 (1H, s, 17α-H), 4.57 (1H, t, J = 5.2 Hz, 19-OH), 4.49 (1H, dd, J = 9.9, 6.1 Hz, 15α-H), 4.38 (1H, s, 17β-H), 3.99 (1H, dd, J = 9.9, 2.4 Hz, 15β-H), 3.84 (1H, dd, J = 10.9, 5.4 Hz, 19α-H), 3.30 (1H, dd, J = 10.9, 4.9 Hz, 19β-H), 2.74 (1H, td, J = 14.3, 5.5 Hz, 2α-H), 2.49–2.30 (3H, m, 2β-H and 11-H), 2.13 (2H, dd, J = 10.7, 3.8 Hz, 7α-H and 9β-H), 1.99 (1H, td, J = 12.8, 4.7 Hz, 7β-H), 1.88 (1H, ddd, J = 12.6, 5.1, 2.8 Hz, 1α-H), 1.72 (1H, d, J = 4.7 Hz, 1β-H), 1.66–1.37 (3H, m, 5β-H and 6-H), 1.01 (3H, s, 18-H), 0.93 (3H, s, 20-H), 0.86 (9H, s, SiC(CH3)3), 0.16 (3H, s, SiCH3), 0.11 (3H, s, SiCH3); 13C NMR (101 MHz, DMSO-d6) δ 213.2 (3-C), 169.4 (16-C), 147.4 (12-C), 147.1 (8-C), 128.2 (13-C), 108.7 (17-C), 73.9 (15-C), 66.7 (14-C), 64.0 (19-C), 55.8 (9-C), 54.4 (5-C), 54.0 (4-C), 38.5 (10-C), 37.9 (7-C), 37.0 (1-C), 35.5 (2-C), 25.6 (SiC(CH3)3), 25.2 (6-C), 24.3 (11-C), 20.3 (18-C), 17.6 (SiC(CH3)3), 14.7 (20-C), −4.4 (SiCH3), −4.9 (SiCH3); HRMS (ESI) m/z 485.2696 [M + Na]+, calculated for C26H42O5SiNa, 485.2699.
In vitro cancer cell proliferation model
Cell culture. Cancer cell line A549 was maintained in RPMI 1640 culture medium, while cancer cell line MDA-MB-231 was maintained in DMEM culture medium (ATCC, USA), which were supplemented with 10% heat-inactivated FBS and 1% P/S. Cells were incubated at 37 °C in a humidified atmosphere with 5% CO2 (v/v).
Cell proliferation assay (MTT assay). A549 or MDA-MB-231 cells were placed into a 96-well plate at a concentration of 0.8 to 1 × 105 cells per well and incubated overnight at 37 °C and 5% CO2 to allow for cell attachment. Various concentrations of testing compounds were added to the cells and then incubated for another 24 h. Cells treated with DMSO (0.1%) served as the vehicle control. After treatment, the medium was discarded and cells were incubated for 4 h at 37 °C in MTT solution (final concentration 1.0 mg ml−1). The solution was then replaced by 100 μl DMSO to dissolve the violet formazan crystals in intact cells. Absorbance was measured by SpectraMaxR M5 Multi-Mode Microplate Readers (Molecular Devices, USA) at 570 nm. CC50 value was determined from the curve of cell viability at which the concentration caused 50% cell death. Each experiment was independently repeated for 3 times.
In vivo zebrafish toxicity assay
Maintenance of zebrafish and collection of embryos. All the animal experiments were conducted according to the ethical guidelines of Institute of Chinese Medical Science (ICMS), University of Macau and the protocol was approved by ICMS, University of Macau. Transgenic zebrafish Tg(fli1a-EGFP)y1 were provided by the Zebrafish International Research Center (ZIRC, Oregon) and wild-type zebrafish were purchased from a local pet shop. Both strains were maintained as described in the zebrafish handbook. Stocks were maintained in a controlled environment (28.5 °C with a 14 h light/10 h dark cycle) and fed with brine shrimp twice a day. Embryos were collected in the morning and cultured in embryo medium at 28.5 °C. At 24 h post fertilization (hpf), the embryos were dechorionated with tweezers in a Petri dish coated with 1% (w/v) agarose, and then distributed into a 6-well plate with 20–50 embryos per group before drug treatment, depending on the assay.
Morphological observations. 24 hpf embryos were incubated in 3 ml of medium containing different concentrations of the test compounds. Embryos receiving DMSO (0.1–0.3%) were used as a vehicle control. Embryos receiving 300 nM VRI were used as a positive control. After drug treatment for 8, 12, and 24 h, the embryos were anesthetized with freshly made 1% (w/v) tricaine (Sigma-Aldrich, St. Louis, MO) and inspected for viability and morphological changes by using an Olympus Microscope System (IX81 Motorized Inverted Microscope [w/ZDC], IX2 universal control box, X-cite series 120, DP71 CCD camera). Images were captured at magnifications of 40× and 100×.
Abbreviations
| SAR | Structure–activity relationship |
| TBS | tert-Butyldimethylsilyl |
| TBSCl | tert-Butyldimethylsilyl chloride |
| TBSOTf | tert-Tutyldimethylsilyl trifluoromethanesulfonate |
| DMP | Dess–Martin periodinane |
| DMAP | 4-Dimethylaminopyridine |
| TFA | Trifluoroacetic acid |
| TEA | Triethylamine |
| Ac | Acetyl |
| p-TSA | p-Toluene-sulfonic acid |
| TBAF | Tetrabutylammonium fluoride |
| Rt | Room temperature. |
Acknowledgements
This work was supported in part by the Natural Science Foundation of China (30973621), Six Major Talents of Jiangsu Province of China (2014) and the Science and Technology Development Fund of Macau SAR (078/2011/A3).
Notes and references
-
(a) M. K. Gorter, Recl. Trav. Chim. Pays-Bas, 1911, 30, 151 CrossRef;
(b) M. P. Cava, W. R. Chan, L. J. Haynes, L. F. Johnson and B. Weinstein, Tetrahedron, 1962, 18, 397 CrossRef CAS.
-
(a) W.-L. Deng, Zhongcaoyaotongxun, 1978, 27 Search PubMed;
(b) W.-L. Deng, J. Liu and R. Nie, Acta Pharmacol. Sin., 1980, 15, 590 CAS;
(c) T. Zhang, Zhongyaocai, 2000, 23, 366 CAS.
-
(a) R. N. Chakravarti and D. Chakravarti, Indian Med. Gaz., 1951, 86, 96 CAS;
(b) X. Liu, Y. Wang and G. Li, Zhongyaocai, 2003, 26, 135 Search PubMed.
-
(a) X. Jiang, P. Yu, J. Jiang, Z. Zhang, Z. Wang, Z. Yang, Z. Tian, S. C. Wright, J. W. Larrick and Y. Wang, Eur. J. Med. Chem., 2009, 44, 2936 CrossRef CAS PubMed;
(b) Z. Wang, P. Yu, G. Zhang, L. Xu, D. Wang, L. Wang and X. Zeng, Bioorg. Med. Chem., 2010, 18, 4269 CrossRef CAS PubMed;
(c) B. Das, C. Chowdhury, D. Kumar, R. Sen, R. Roy, P. Das and M. Chatterjee, Bioorg. Med. Chem. Lett., 2010, 20, 6947 CrossRef CAS PubMed;
(d) V. Menon and S. Bhat, Nat. Prod. Commun., 2010, 5, 717 CAS;
(e) U. Sirion, S. Kasemsook, K. Suksen, P. Piyachaturawat, A. Suksamrarn and R. Saeeneg, Bioorg. Med. Chem. Lett., 2012, 22, 49 CrossRef CAS PubMed.
- Z. Liu, W.-K. Law, D. Wang, X. Nie, D. Sheng, G. Song, K. Guo, P. Wei, P. Ouyang, C.-W. Wong and G.-C. Zhou, RSC Adv., 2014, 4, 13533 RSC.
-
(a) R. S. Chang, L. Ding, G. Q. Chen, Q. C. Pan, Z. L. Zhao and K. M. Smith, Proc. Soc. Exp. Biol. Med., 1991, 197, 59 CrossRef CAS;
(b) A. Basak, S. Cooper, A. G. Roberge, U. K. Banik, M. Chretien and N. G. Seidah, Biochem. J., 1999, 338, 107 CrossRef CAS.
-
(a) J. Rihel, D. A. Prober, A. Arvanites, K. Lam, S. Zimmerman, S. Jang, S. J. Haggarty, D. Kokel, L. L. Rubin, R. T. Peterson and A. F. Schier, Science, 2010, 327, 348 CrossRef CAS PubMed;
(b) D. Kokel, J. Bryan, C. Laggner, R. White, C. Y. Cheung, R. Mateus, D. Healey, S. Kim, A. A. Werdich, S. J. Haggerty, C. A. Macrae, B. Shoichet and R. T. Peterson, Nat. Chem. Biol., 2010, 6, 231 CrossRef CAS PubMed.
- I. K. Lam, D. Alex, Y.-H. Wang, P. Liu, A.-L. Liu, G.-H. Du and S. M. Y. Lee, Mol. Nutr. Food Res., 2012, 56, 945 CAS.
-
(a) Z. H. Li, D. Alex, S. O. Siu, I. K. Chu, J. Renn, C. Winkler, S. Lou, S. K. Tsui, H. Y. Zhao, W. R. Yan, G. B. Mahady, G. H. Li, Y. W. Kwan, Y. T. Wang and S. M. Lee, Mol. BioSyst., 2011, 7, 2128 RSC;
(b) G. Hu, S. O. Siu, S. Li, I. K. Chu, Y. W. Kwan, S. W. Chan, G. P. Leung, R. Yan and S. M. Lee, Xenobiotica, 2012, 42, 294 CrossRef CAS PubMed.
- R. A. Kumar, K. Sridevi, N. V. Kumar, S. Naduri and S. Rajagopal, J. Ethnopharmacol., 2004, 92, 291 CrossRef CAS PubMed.
- H. T. Gao, B. L. Wang and W. D. Z. Li, Tetrahedron, 2014, 70, 9436 CrossRef CAS PubMed.
- X. Xue, L. Li, Y. Song, Y. Lu and J. Li, China Pat., CN 103588738 A, 2014, vol. 2, 07.
- S. R. Jada, G. S. Subur, C. Matthews, A. S. Hamzah, N. H. Lajis, M. S. Saad, M. F. G. Stevensc and J. Stanslas, Phytochemicals, 2007, 68, 904 CrossRef CAS PubMed.
- U. Sirion, S. Kasemsook, K. Suksen, P. Piyachaturawat, A. Suksamrarn and R. Saeeng, Bioorg. Med. Chem. Lett., 2012, 22, 49 CrossRef CAS PubMed.
- S.-M. Wang, Y.-B. Zhang, H.-M. Liu, G.-B. Yu and K.-R. Wang, Steroids, 2007, 72, 26 CrossRef CAS PubMed.
- X. He, X. Zeng, H. Hu and Y. Wu, J. Mol. Catal. B: Enzym., 2010, 62, 242 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of compounds 4a/4b to 25a/25b. See DOI: 10.1039/c5ra00090d |
| ‡ These authors contributed equally to this paper. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.