
Largely improved electromechanical properties of thermoplastic polyurethane dielectric elastomer by carbon nanospheres
Abstract
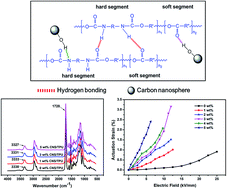
Carbon nanospheres (CNS) were used as a new conductive filler to improve the electromechanical properties of a thermoplastic polyurethane (TPU) dielectric elastomer (DE). The results showed that CNS with many hydroxyl groups can form hydrogen bonds with TPU molecules, leading to a good dispersion of CNS in the TPU matrix and an improved tensile strength of CNS/TPU composites. More interestingly, CNS disrupted the crystallization of TPU, resulting in the decrease in elastic modulus and hysteresis loss of the composites. The dielectric constant at 1000 Hz increased from 7.1 for pure TPU to 137.3 for the composite with 5 wt% of CNS. The great increase in dielectric constant and the decrease in elastic modulus result in the largely improved actuation strain at low electric field of CNS/TPU composites. In addition, all the as-prepared CNS/TPU composites have a low dielectric loss (<1) at 1000 Hz. Our study provides a simple and effective way to obtain CNS/TPU DE with good mechanical strength and largely improved actuation performance at low electric field.
 Please wait while we load your content...
Please wait while we load your content...

