
Rational design of 5-HT6R ligands using a bioisosteric strategy: synthesis, biological evaluation and molecular modelling†
Abstract
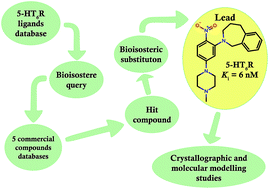
A bioisosteric strategy was successfully implemented with a screening protocol for new, potent 5-HT6R ligands. Initially, 2-[5-(4-methylpiperazin-1-yl)-2-nitrophenyl]-1,2,3,4-tetrahydroisoquinoline (9) was found in commercial databases using a bioisosteric query (screening 5-HT6R Ki = 128 nM). Then, the hit compound was bioisosterically modified (ring alteration) leading to a novel, high affinity (Ki = 6 nM) 5-HT6R ligand (10). Extensive docking studies followed by structural interaction fingerprint analysis supported by single-crystal X-ray structures of the investigated ligands suggest different binding modes with 5-HT6R models for compounds with varying activity. An alternative anchoring point for protonated amine (D7.36) that has not been previously reported was identified.
 Please wait while we load your content...
Please wait while we load your content...

