
Scaffold-hopping and hybridization based design and building block strategic synthesis of pyridine-annulated purines: discovery of novel apoptotic anticancer agents†
Abstract
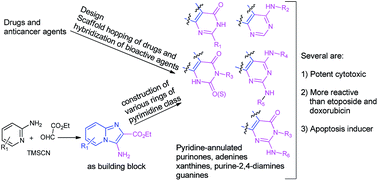
A set of novel pyridine-annulated analogs of purinones, adenines and their oxo/thio congeners, xanthines, guanines, and purine-2,4-diamines as potential anticancer agents was considered based on the scaffold-hopping and hybridization of known anticancer agents/drugs, purine derivatives and our recently developed imidazo-pyridine derivatives. Towards the synthesis of these compounds, a new approach involving a convenient preparation of 3-amino-2-carboxyethyl substituted imidazo[1,2-a]-pyridine and its use as a building block for the construction of fused rings was developed. The approach enabled the preparation of a number of compounds with relevant substitutions for each class. Several of pyridine-annulated adenine and its oxo/thio analogs, xanthine and purine-2,4-diamine were found to possess significant anticancer activities in kidney cancer cells and relatively less cytotoxicity to normal cells. They were relatively more active than the anticancer drugs etoposide and doxorubicin. A representative pyridine-annulated adenine derivative compound(22) was found to exert significant apoptosis.
 Please wait while we load your content...
Please wait while we load your content...

