
The preparation of Fe2O3 nanoparticles by liquid phase-based ultrasonic-assisted method and its application as enzyme-free sensor for the detection of H2O2
Chen Hao*a,
Feng Fenga,
Xiaohong Wang*a,
Min Zhoua,
Yutao Zhaob,
Cunwang Gec and
Kun Wanga
aSchool of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang, Jiangsu 212013, China. E-mail: chhao@ujs.edu.cn; xhwang@ujs.edu.cn; Fax: +86 511 88791800; Tel: +86 511 88791800
bSchool of Material Science & Engineering, Jiangsu University, Zhenjiang, Jiangsu 212013, China
cSchool of Chemistry and Chemical Engineering, Nantong University, Nantong, Jiangsu 226019, China
First published on 16th February 2015
Abstract
Iron oxide nanoparticles with high electrocatalytic activity for hydrogen peroxide were developed by liquid phase-based ultrasonic-assisted method using sodium lignosulphonate as surfactant. The influence of the different preparation conditions including addition of sodium lignosulfonate (SLS) and calcining temperature was investigated by X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and Brunauer–Emmett–Teller (BET) specific surface area. Then, the as-prepared Fe2O3 with graphene (G) was further fixed on the surface of glassy carbon electrode (GCE) using chitosan (CS) as a crosslinking agent. The electrochemical properties of the prepared G-Fe2O3-NPS-CS/GCE senor were estimated by cyclic voltammetry and chronoamperometry. Finally, the G-Fe2O3-NPS-CS/GCE (1.0 g SLS, calcined 400 °C) senor showed an excellent electrocatalytic activity towards hydrogen peroxide, which displayed high sensitivity (385.59 μA mM−1 cm−2), wide detection range (0.5–7800 μM), low detection limit (0.5 μM) and a fast response time less than 2 s. Furthermore, the sensor also exhibited good anti-interference for ascorbic acid and uric acid, excellent repeatability and long-term stability. These results indicated that the G-Fe2O3-NPS-CS/GCE (1.0 g SLS, calcined 400 °C) senor held great potential for the detection of hydrogen peroxide.
Introduction
Nanomaterials have become one of the most active research orientations in the areas of physics, chemistry, and engineering since 1990, due to the small particle size, large specific surface area, high surface energy and unique surface effect, small-size effect, and macroscopical quanta tunnel effect, etc.1–4 In the past several decades, various types of nanomaterials such as Au, Ag, TiO2, SiO2, and ZnO etc. have been widely applied as photocatalyst, functional ceramics, sensor, solar cell and biology functional material.5–9 Among the various kinds of nanomaterials, iron oxide as an important oxide has been studied for a wide range of applications because it is environmentally friendly, non-toxic, heat-resistant, and corrosion-resistant material.10,11 At the same time, because of the low density, large surface, high stability, and remarkable sensitivity of the conductivity to temperature, humidity and concentration, the Fe2O3 semiconductor shows a broad and good prospect for application in the field of sensing materials.On the other hand, the reliable and rapid determination of hydrogen peroxide (H2O2) is of great importance in biology and chemistry fields because it is not only used as an important oxidizing agent in food and chemical industries, but also widely used as a mediator in food, pharmaceutical, clinical, industrial and environmental analysis.12–15 Many techniques have been successfully used for the detection of H2O2, such as titrimetry, spectrophotometry, fluorescence, chemiluminescene and electrochemistry. Compared with other detection methods, electrochemistry has attracted more and more interest of researchers, due to the convenience, high sensitivity, and excellent precision of the technique.16–18 Some electrochemical sensors with a high sensitivity and specificity for detection of H2O2, such as NF/CAT/MWCNTs-COOH/Cys-AuNPs/GC, CAT/MgO-NPs/CPE, GC/MWCNT-NiO/CAT and GC/MWCNTs/[bmim][PF6]/CAT have been successfully developed.19–22 Most of the above mentioned electrochemical sensors were constructed based on enzymes or proteins. Enzyme-modified electrochemical sensors can achieve high sensitivity and excellent selectivity, however, there are many defects of enzyme-based electrochemical sensors, such as instability, limited lifetime, high cost and complicated modification procedure. Its activity, meanwhile, can be easily affected by temperature, pH value and oxygen.23–27 In comparison with the enzyme-modified sensors, the enzyme-free sensor's research becomes very meaningful duo to their long lifetime, high environmental suitability and high durability.
Iron oxide has been a good choice to be used as substrate for enzyme-free sensors due to its good chemical stability, low-cost and narrow band gap (−2.1 eV).28 It is well-known that the excellent performance of the material depends not only on the characteristic of the material itself, but also on the size, crystallinity and morphology of the material particle.29 There are always some problems like agglomeration of particles, large particle size, etc. exist during the preparation of iron oxide. Therefore, control of the size and morphology of Fe2O3 has attracted more and more attention of researchers. Up to now, some Fe2O3 particles with excellent properties have been synthesized through various methods with the assistance of different surfactants.30–32
In this paper, a technically simple and cost-effective ultrasonic method for synthesizing Fe2O3 nanoparticle in the presence of SLS has been developed in order to obtain the desired sensor materials. Then, the as-prepared Fe2O3 nanoparticle and graphene, were immobilized onto the GCE surface by chitosan. Cyclic voltammetry and chronoamperometry are used to determine the electrochemical properties of the sensor. The resulting sensor exhibited high sensitivity, a wide linear range and a lower detection limit for hydrogen peroxide.
Experimental
Materials
Ferric chloride, carbamide, potassium permanganate, flake graphite, hydrazine hydrate and hydrogen peroxide were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Concentrated hydrochloric acid (36–38%, m m−1) and concentrated sulfuric acid (98%, m m−1) were purchased from Yangzhou Hubao Chemical Reagent Co., Ltd. (Yangzhou, China). SLS of industrial grade was bought from Fei Wong Xinyi Chemical Co., Ltd (Xuzhou, China). All other reagents were of analytical grade and used without further purification. 0.2% (m m−1) chitosan solution was prepared by addition of chitosan in 1% (m m−1) acetic acid solution. 0.1 M of pH = 7.4 phosphate buffer solution (PBS) was adjusted by 0.1 M K2HPO4 and KH2PO4. All as-prepared solution were stored at 4 °C when not in use.Preparation of Fe2O3 nanoparticle and graphene
Fe2O3 nanoparticles were synthesized by liquid phase-based ultrasonic-assisted method in the following way: 4.06 g FeCl3·6H2O was dissolved in 250 mL double-distilled water with certain amount of SLS and the mixed solution was sonicated in the water bath of 85 °C until a relatively steady solution system was formed. Then a 50 mL solution of 1.2 M (NH2)2CO was added to the system drop by drop, and the reaction system was refluxed for 4 h. The resultant solids were collected by filtration and repeated washing. After that, the solid was dried in oven at 60 °C, and then calcined at 300 °C, 400 °C, 500 °C and 600 °C in furnace for 2 h, respectively. The added amounts of SLS were 0, 0.5, 1.0, 1.5, 2.0 g, respectively.Graphite oxide was prepared using flake graphite by a modified Hummer's method:33 first, graphite powder was oxidized by KMnO4/NaNO3 and H2SO4, and the graphite oxide was exfoliated into graphene oxide by a KQ-250 ultrasonic reactor (Kunshan, China, P = 250 W, f = 40 kHz). Subsequently, the unexfoliated graphite oxide is separated by centrifugation at 2000 rpm for 10 min. Finally, the obtained back suspension of graphene oxide is reduced into graphene using hydrazine. The graphener was washed for several times and stored at 4 °C.
Preparation of the modified electrodes
The preparation of the G-Fe2O3-NPS-CS/GCE modified electrode is described as follows: prior to use, the bare glassy carbon electrode (GCE, 4 mm in diameter) was carefully polished to a mirrorlike surface with emery paper and 1.0 μm, 0.5 μm alumina slurry, respectively. Then the polished electrode was rinsed ultrasonically in acetone, absolute ethanol, deionized water for 3 min, respectively. The electrode was activated using repeatedly cyclic voltammetry scanned from −1.0 to 1.0 V at 50 mV s−1 in 1.0 M H2SO4 until a stable voltammogram was obtained. To prepare the modified composite, 3 mg of Fe2O3 and 2 mg of graphene are mixed in 0.2% (m m−1) chitosan solution, and this mixture was sonicated for 30 min to acquire a homogeneous and well-dispersed black suspension. Whereafter, 3 μL of the suspension was smeared evenly onto surface of the GCE electrode. In order to obtain a more uniform modified films, the as-prepared electrode was slow dried at 4 °C. The obtained G-Fe2O3-NPS-CS/GCE modified electrodes was stored at 4 °C. Meanwhile, the Fe2O3-NPS-CS/GCE modified electrode was also prepared. Iron oxide and graphene easily fall off from the surface of electrode without the adhesive force of chitosan (CS) for the G-Fe2O3-NPS/GCE modified electrode. Therefore, the G-Fe2O3-NPS/GCE has not been discussed in this work. The G-Fe2O3-NPS-CS/GCE (1.0 g SLS, calcined 400 °C) are denoted as G-Fe2O3-CS/GCE-1-400.Characterizations
Thermogravimetric/differential scanning calorimetry (TG/DSC) analysis was performed using a integrated thermal analyzer (STA449C, NETZSCH) under a nitrogen atmosphere with a heating rate of 5 °C min−1 from 25 °C to 1000 °C. FT-IR spectra of the powders (as pellets in KBr) were recorded using Fourier transform infrared spectroscopy (FT-IR, Nicolet, AVATAR-370MCT) in the range of 4000–400 cm−1. The XRD patterns of the Fe2O3 nanoparticles were recorded using a Bruker D8 Advance X-ray diffractometer at a voltage of 40 kV and current of 40 mA with Cu Kα radiation (λ = 0.15406 nm). The specific surface area and pore size data were obtained by surface area and porosity analyzer (NOVA-2000e). The morphology of the nanoparticles was examined with the Field Emission Scanning Electron Microscope (FE-SEM, S4800, HI-9140-0006). All electrochemical experiments were performed by a CHI 660C electrochemical workstation (Chen Hua Instrumental Corporation, Shanghai, China). A conventional three-electrode system (including the modified GCE as the working electrode, a platinum foil as the counter electrode and a saturated calomel electrode (SCE) as the reference) was employed to test electrochemical property of modified electrode.Results and discussion
TG–DSC measurement was performed to investigate the decomposition behavior and the phase transition temperatures of the as-synthesized precursor (FeOOH) containing 1.0 g SLS. The results are exhibited in Fig. 1. | ||
| Fig. 1 DSC–TGA curve for α-FeOOH precursor containing SLS. | ||
The curve of TG shows that the first weight loss was about 3.87% in temperature range of 25 °C to 100 °C, which is attributed to the dehydration of the physical adsorption on the sample surface. The relatively large weight loss of 21.47% occurred from 100 °C to 480 °C, and the corresponding exothermic peak was observed at 298.9 °C in the DSC curve. This is because of the decomposition of the α-FeOOH34 and SLS at high temperature. The third mass loss of 1.44% from 480 °C to 820 °C is attributed to the continued decomposition of SLS surrounded by Fe2O3. No obvious weight loss over 820 °C. Subsequently, the DSC curve shows that the small exothermic peak at 599.6 °C is mainly due to the phase transformation from γ-Fe2O3 to α-Fe2O3.35
The FT-IR spectra as an important analytical technique was used to evaluate the structural changes of the different samples shown in Fig. 2. It can be found that there are large numbers of functional groups of SLS among the as-prepared α-FeOOH containing 1.0 g SLS. The results indicate that the SLS has been successfully embedded in the α-FeOOH precursor. Accompanied by the thermal decomposition of α-FeOOH precursor into Fe2O3, the characteristic peaks of SLS almost all disappeared due to the volatilization and decomposition of SLS. The two obvious absorption peaks at 475 cm−1 and 442 cm−1 are attributed to the vibration of the Fe3+–O2−.
 | ||
| Fig. 2 FT-IR spectra of sodium lignosulphonate, α-FeOOH and Fe2O3. | ||
Fig. 3A shows the XRD patterns of the as-prepared samples with varied additions of SLS calcined at 400 °C. The curve with the narrowest and sharpest peaks in Fig. 3A indicates the best crystallized Fe2O3 for the addition of no SLS. All XRD peaks at 2θ = 24.1°(012), 33.2°(104), 35.6°(110), 40.9°(113), 49.5°(024), 54.1°(116), 57.4°(018), 62.5°(214), 64.0°(300) and 72.0°(1010) can be clearly indexed to the rhombohedral phase of hematite (JCPDF no. 33-0664). No characteristic peaks of any other impurities are detected. The diffraction peak be gradually weakened at (012), (104), (113), (024), (214), (300) and (1010) with the increasement of SLS. These characteristic peaks almost disappear for the addition of 2.0 g SLS. Meanwhile, it is obvious to compare the JCPDF cards that we can find the transformation of Fe2O3 crystalline structure from the rhombohedral symmetry (JCPDS no. 33-0664) to Maghemite (JCPDS no. 39-1346) with the increasing SLS content. In addition, the influence of different calcination temperature was also discussed, as shown in Fig. 3B. The Fe2O3 samples calcined at 300 °C showed wider and weaker diffraction peaks, and displayed poorer crystallization than the other Fe2O3 samples. As expected, the diffraction peaks' intensity of Fe2O3 constantly reinforced with an ever-increasing calcination temperature, accompanied with the gradual growth of Fe2O3 nanoparticles. γ-Fe2O3 was successfully transformed to α-Fe2O3 with the increase in the annealing temperature. The average crystallite dimension (ACD) of all samples was estimated through using Scherrer formula, D = 0.9λ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ, where D is the average crystalline size, λ is the wavelength of Cu Kα (λ = 0.15406 nm), β is the full width at half maximum of the diffraction peaks, and θ is the Bragg's angle. the results of the computation are shown in Table 1.
cosθ, where D is the average crystalline size, λ is the wavelength of Cu Kα (λ = 0.15406 nm), β is the full width at half maximum of the diffraction peaks, and θ is the Bragg's angle. the results of the computation are shown in Table 1.
 | ||
| Fig. 3 (A) XRD patterns of as-prepared Fe2O3 samples with different additions of SLS calcined at 400 °C. (B) XRD patterns of Fe2O3 samples prepared with 1.0 g SLS calcined at 300 °C, 400 °C, 500 °C and 600 °C. | ||
| SLS/g | T/°C | ACD/nm | BET/m2 g−1 | SLS/g | T/°C | ACD/nm | BET/m2 g−1 |
|---|---|---|---|---|---|---|---|
| 0.0 | 400 | 32.03 | 8.208 | 2.0 | 400 | 9.26 | 46.311 |
| 0.5 | 400 | 26.78 | 27.430 | 1.0 | 300 | 76.196 | |
| 1.0 | 400 | 21.86 | 81.319 | 1.0 | 500 | 32.13 | 21.367 |
| 1.5 | 400 | 10.06 | 71.912 | 1.0 | 600 | 36.21 | 11.709 |
The morphology of the as-prepared Fe2O3 nanoparticles are shown in Fig. 4. The changes in morphology before and after the addition of sodium lignosulfonate are noticeable. The pure sample without surface modification is shown in Fig. 4A. It can be clearly seen that the particle of Fe2O3 is bulky compared to the modified Fe2O3 samples, and the primary particle size is between 200 and 500 nm. With the increase of SLS content, the size of the particles of iron oxide decreased rapidly. As shown in the Fig. 4C, the as-prepared Fe2O3 samples are composed of fine and uniform particles with the addition of 1.0 g SLS. Fig. 4F is a higher magnification of Fig. 4C, the synthesized Fe2O3 nanoparticles showed smallest grain size (ca. 20 nm in diameter) and abundant porous nanostructure. Such structure exhibits a high-developed surface area. Interestingly, the grain size is raised to 30 nm when the quality of SLS increased to 1.5 g, as shown in Fig. 4D. Meanwhile, it can be clearly observed that severe agglomeration has occurred between particles, which indicates that an excessive increase in the addition amount of SLS is detrimental to the preparation of Fe2O3. The morphology of the Fe2O3 sample added 1.0 g SLS to the reaction system without ultrasound was also investigated (Fig. 4E), The results suggest that the growth of Fe2O3 is nearly uncontrolled, and the particle size of Fe2O3 is even larger than 500 nm. The calcining temperature affects the final quality and performance of the powders, so it is essential to discuss and research the influence of the temperature on the preparation of nanomaterials. The typical SEM image of Fe2O3 annealed at 300 °C is displayed in Fig. 5A. The heavily agglomerated particles and XRD patterns show that the as-prepared precursor is not successfully transformed to Fe2O3 crystal under the lower calcination temperature. It can be observed that the particle size show an increasing trend with increase in temperature, and change from smaller than 30 nm at 400 °C (Fig. 4C) to about 60 nm at 500 °C (Fig. 5B). The results showed that the surfactant and the calcining temperature have great influence on the particle size of Fe2O3.
 | ||
| Fig. 4 SEM images of Fe2O3 samples with different additions of SLS calcined at 400 °C: (A) 0 g, (B) 0.5 g, (C) 1.0 g, and (D) 1.5 g; (E) SEM images of Fe2O3 samples adding 1.0 g SLS without assistance of ultrasound; (F) high magnification of (C). | ||
 | ||
| Fig. 5 SEM images of Fe2O3 samples with the addition of SLS is 1.0 g calcined at 300 °C (A), 500 °C (B). | ||
Nitrogen isotherm adsorption–desorption experiment was carried out to examine characteristics of the specific surface areas of the as-prepared Fe2O3 under different conditions, the results are shown in Table 1. Without the addition of SLS, the Fe2O3 nanoparticles show small specific surface area of 8.208 m2 g−1. After that, the specific surface areas of the Fe2O3 is consistently increasing with the added amount of SLS, and an maximum specific surface areas (81.319 m2 g−1) was obtained when the SLS is added to 1.0 g. The Fe2O3 calcined at 400 °C display highest specific surface areas than those of 300 °C, 500 °C and 600 °C. By comparing the analysis results of XRD and SEM, we can find that the specific surface areas increases with the decreasing of particle diameter because of the surface effect.
According to the SEM, XRD and BET results mentioned above, the impacts of SLS, ultrasonic and calcining temperature on the formation of Fe2O3 can be described as follows: first, from the FTIR spectrograms it is observed that sodium lignosulphonate contains a large amount of carboxylic (3437 cm−1) and sulfonic (1044 cm−1 and 621 cm−1) groups.36,37 After adding the surfactant SLS to ferric chloride solution, SLS produces the negatively-charged sulfonic and carboxylic groups that could form a covalent bond with Fe3+ by electrostatic interactions.38 The appropriate SLS concentration leads to the lower Fe3+ concentration, which will decrease the nucleation rate of FeOOH. Meanwhile, these negatively charged groups can be absorbed on the crystal nucleus surface under low nucleation rate. These behaviors will help inhibit the crystal nucleus growth and agglomeration. However, when the excessive addition of SLS, these negatively-charged groups might aggregate with other positively-charged groups by electrostatic interactions, and lose its function to suppress the crystal nucleus growth and agglomeration. On the other hand, higher ultrasonic irradiation can generate more nucleation which leads to smaller grain size and more dispersed particles.39 The influence of temperature, we can speculate that the decomposition of FeOOH gradually increased with the increase in calcination temperature, and the high decomposition rate will speed up the crystal growth. Finally, the Fe2O3 grain size also continuously increased with the rapid crystal growth rate.
TEM images of G and G-Fe2O3 shown in Fig. 6 provide strong evidence that Fe2O3 has been immobilized on the surface of G by a bridge constituted of chitosan (Black spots or gray dots in Fig. 6B). It can be seen from Fig. 6A that the G displays typical semitransparent flakelike and some crumpled shapes, which is attributed to the thin thickness and large surface area of G. This is an advantage for electron transfer on the surface of the electrode. Fe2O3 has been tightly stuck to the wrinkled G surface using the chitosan, as shown in Fig. 6B. The average diameter of Fe2O3 was estimated to be about 20 nm, which is roughly correspondence with the XRD and SEM analysis results.
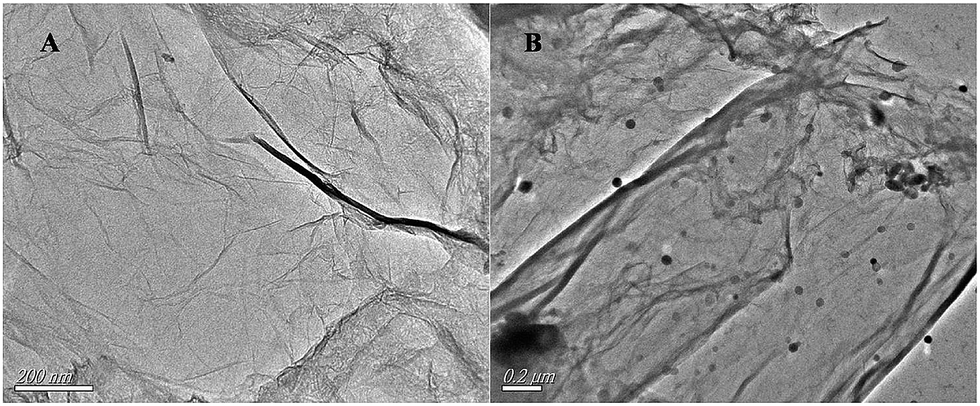 | ||
| Fig. 6 TEM images of G (A) and G-Fe2O3-NPS-CS (B). | ||
The electrochemical properties of the as-prepared Fe2O3 nanoparticles modified GCE electrode was detected using cyclic voltammetry in [Fe(CN)6]3−/4− solution with the scan rate 50 mV s−1 at room temperature. First the bare GCE, Fe2O3-NPS-CS/GCE, G-Fe2O3-NPS-CS/GCE electrodes were tested in a solution of 5 mM [Fe(CN)6]3−/4− containing 0.1 M KCl in the range from −0.1 to 0.6 V, as shown in Fig. 7A. Only the Fe2O3 modified GCE electrode shows a low pair of redox peaks compared with bare GCE electrode. This could be due to iron oxide semiconductor hinder the electronic transmission on the surface of GCE. In order to improve electrode properties, graphene was introduced into Fe2O3 modified GCE. It is clear that the introduction of graphene gives rise to a sharp increase of the peak current. The cyclic voltammogram of the above three electrodes in 0.1 M PBS of pH 7.4 in the range from −0.8 to 0.8 V with the presence and the absence of 5 mM hydrogen peroxide was shown in Fig. 7B.
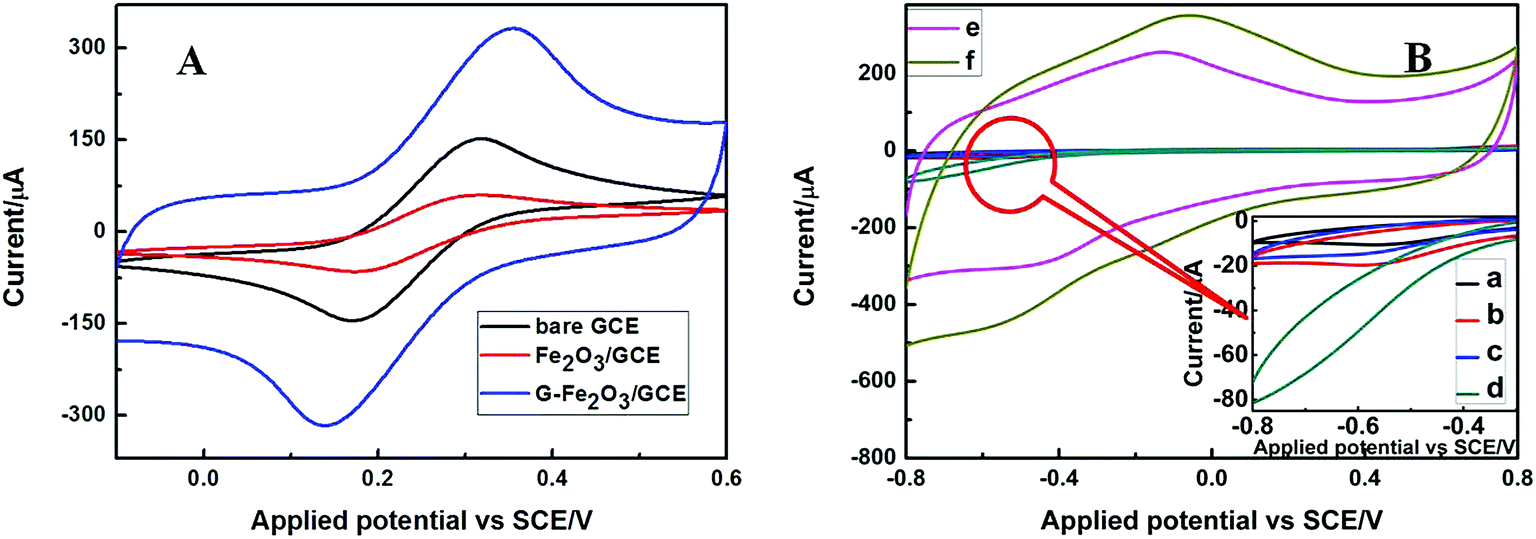 | ||
| Fig. 7 (A) CVs of the bare GCE, Fe2O3-NPS-CS/GCE and G-Fe2O3-NPS-CS/GCE in 5 mM [Fe(CN)6]3−/4− solution (1:1, molar ratio) with 0.1 M KCl. (B) CVs of bare GCE (a and b), Fe2O3-NPS-CS/GCE (c and d) and G-Fe2O3-NPS-CS/GCE (e and f) in the absence (a, c and e) and presence (b, d and f) of 5 mM H2O2 in 0.1 M PBS. Inset: local amplification of the selected area. | ||
At the bare electrode in the absence (curve a) and presence (curve b) of H2O2, no obvious current responses were observed. The Fe2O3 modified electrode showed an obvious current response at about −0.38 V in the presence of H2O2 (curve d). It can clearly be seen that, after adding the graphene into the modified electrode (G-Fe2O3-NPS-CS/GCE), the current response of the electrode dramatically increases. The results display that the electron transfer rate of the G-Fe2O3-NPS-CS/GCE is much higher than that of the Fe2O3-NPS-CS/GCE. Meanwhile, the G-Fe2O3-NPS-CS/GCE modified electrode also show excellent sensitivity to H2O2 (curve f), which could be attributed to electrochemical signal amplification through the good conductivity and the large surface area of the graphene. It also indicated that the G-Fe2O3-NPS-CS/GCE composite sensor was developed successfully.
The electrochemical properties of the as-prepared Fe2O3 nanoparticles with different addition amount of SLS calcined under 400 °C are shown in Fig. 8. Fig. 8A depicts the cyclic voltammogram of these modified electrodes in 5 mM [Fe(CN)6]3−/4− containing 0.1 M KCl, it is found that all electrodes exhibited a pair of obvious redox peaks, and the current response gradually increased with the addition of SLS. The value of redox peak current reached the maximum at the addition of 1.0 g SLS. However, the redox peak current was accompanied by a dramatical decrease with a further increase of SLS. Fig. 8B shows the CVs of the Fe2O3 modified electrode in 0.1 M PBS (pH 7.4) toward the reduction of 5 mM H2O2. A similar phenomenon in which the current response increased with the addition of SLS was observed for these electrodes. After added 1.0 g, the sensitivity of the sensor to hydrogen peroxide started to decline gradually with the continuous addition of SLS. Therefore, we can come to the conclusion that it is detrimental for preparation of senor due to an excessive addition of SLS.
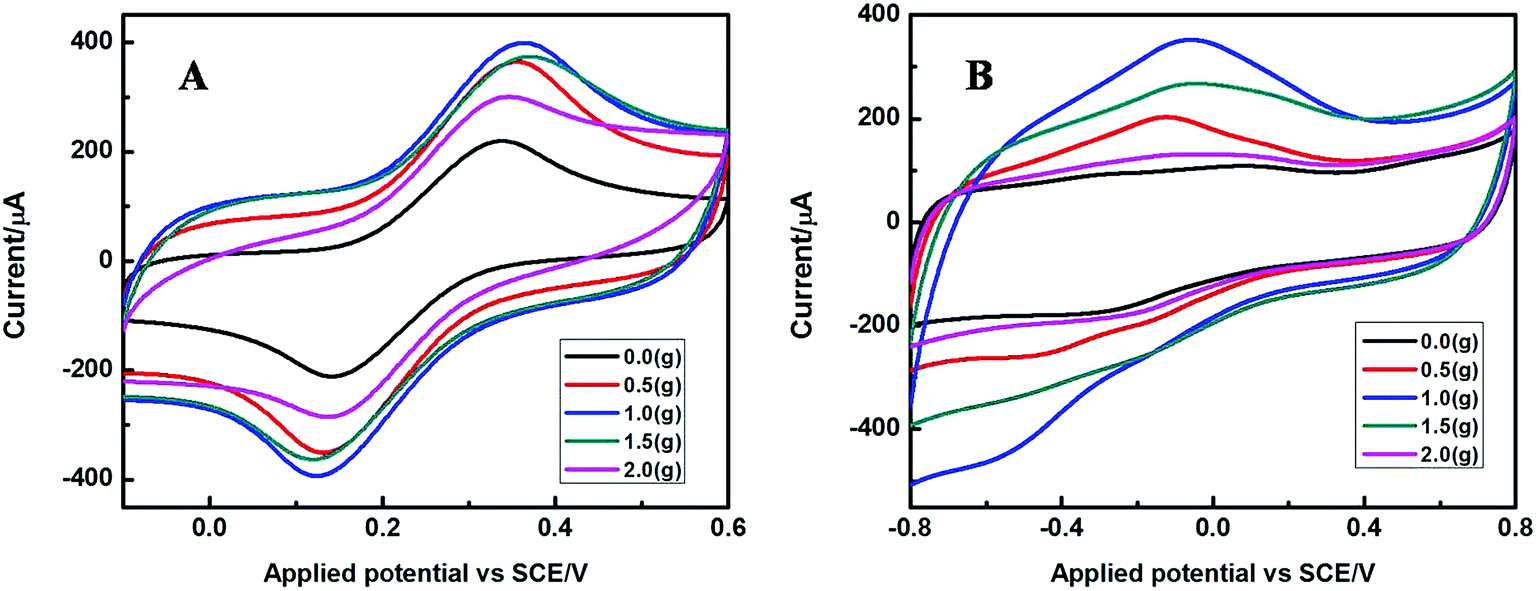 | ||
| Fig. 8 CVs of G-Fe2O3-NPS-CS/GCE in 5 mM [Fe(CN)6]3−/4− solution (1:1, molar ratio) with 0.1 M KCl (A) and in 0.1 M PBS (pH = 7.4) containing 5 mM H2O2 (B), the Fe2O3-NPS used in this experiment were calcined at 400 °C, and with the addition of 0 g, 0.5 g, 1.0 g, 1.5 g, and 2.0 g SLS. | ||
Fig. 9 shows the cyclic voltammograms of different G/Fe2O3-NPS/CS/CPEs in which the Fe2O3-NPS calcined at various temperatures. The electron transfer rate between the G-Fe2O3 composite film and electrode surface was studied in 5 mM [Fe(CN)6]3−/4− containing 0.1 M KCl, as shown in Fig. 9A. When the Fe2O3 was calcined at 400 °C, the electron transfer rate of the G/Fe2O3-NPS/CS/CPE is faster than that of the others. Electrochemical properties of the as-prepared modified electrodes are further detected in the presence of 5 mM H2O2 in 0.1 M PBS (pH = 7.4). It can be seen from Fig. 9B that the current response of the G/Fe2O3-NPS/CS/CPE modified electrode (Fe2O3 calcined at 400 °C) at the −0.38 V was improved 10%, 20% and 30% respectively, compared with other three electrodes (Fe2O3 calcined at 300 °C, 500 °C and 600 °C). Apparently, the above presented results indicated that calcination temperature is an important influencing factor during the preparation process of Fe2O3. Combining with the analyses of XRD, SEM and CVs, it can be found that the sensitivity of the H2O2 sensor increases with decreasing of Fe2O3 nanoparticle size. We speculate that this may be due to the intense surface force field, lack of particle coordination and elevated defects of the small particles with larger specific surface area, which are beneficial to the absorbance of H2O2 in solution.
 | ||
| Fig. 9 CVs of G-Fe2O3-NPS-CS/GCE in 5 mM [Fe(CN)6]3−/4− solution (1:1, molar ratio) with 0.1 M KCl (A) and in 0.1 M PBS (pH = 7.4) with 5 mM H2O2 (B), the Fe2O3-NPS used in this experiment were doped with 1.0 g SLS, and calcined at 300 °C, 400 °C, 500 °C and 600 °C. | ||
Finally, the sensitivity of G-Fe2O3-CS/GCE-1-400 is estimated by chronoamperometry measuring the current response with the gradually addition of H2O2 to 0.1 M BPS of pH 7.4 at fixed potential of −0.38 V. As shown in the Fig. 10 the sensor showed a fast amperometric response time less than 2 s, which is greatly shortened compared to the previous literature reported for other H2O2 sensors.13,40,41 Meanwhile, with the increasing of H2O2 concentration, the amperometric response of the G-Fe2O3-CS/GCE-1-400 electrode increased linearly (Table 2). The linear regression equation was expressed as: Ip (A) = −53.02 − 48.43[H2O2] (mM) in the wide calibration range from 0.5 to 7800 μM with a correlation coefficient of 0.999. A low detection limit of 0.5 μM is estimated at the signal to noise of 3. The sensitivity of the G-Fe2O3-CS/GCE-1-400 electrode is calculated to be 385.59 (μA mM−1 cm−2) by the slope of the linear regression curve. The as-prepared electrode exhibits high sensitivity and a low detection limit compared to other modified electrodes, such as the Ag-nanofibrous membrane/GCE,6 heme protein-SWCNT-CTAB electrodes42 and CTAB-SAMN/CPE.43 These results demonstrate that H2O2 could be easily detected used as-prepared modified electrode.
 | ||
| Fig. 10 Amperometric response of sensor to H2O2 concentration in 0.1 M pH = 7.4 PBS at −0.38 V. Inset: linear plot. | ||
| Electrode materials | LR (μM) | Sensitivity | DL (μM) | RT | Reference |
|---|---|---|---|---|---|
| Ag-nanofibrous membrane/GCE | 10–16500 |
157 μA mM−1 cm−2 | 4 | 6 | |
| Au/graphene/HRP/CS/GCE | 5.0–5130 | 1.7 | <3 s | 13 | |
| AgNPs/Ox-pTTBA/MWCNT | 10–260 | 0.24 | <5 s | 40 | |
| Hb/HMS-modified GCEs | 0.4–6.0 | 1.86 × 10−3 | <5 s | 41 | |
| α-Fe2O3NR arrays | 0.2–5000 | 135.36 μA mM−1 cm−2 | 0.1 | 42 | |
| CTAB-SAMN/CPE | 10–1500 | 58 μA mM−1 cm−2 | 2.78 | 43 | |
| G-Fe2O3-NPS-CS/GCE | 0.5–7800 | 385.59 μA mM−1 cm−2 | 0.5 | <2 s | This work |
Anti-interference test, being the crucial influence factor, can never be neglected in the determination of any objects by electrochemical method. Therefore, the influence of common interfering substances such as ascorbic (AA) and uric acid (UA) was studied by amperometric method. The results is shown in the Fig. 11, the modified electrode show a weak current response to the addition of 5 mM AA and UA with the 0.1 M BPS of pH 7.4 at fixed potential of −0.38 V. This illustrates the proposed sensor well prevented the influence of interfering substances. Stability and repeatability studies were conducted to further detection the electrochemical performance of G-Fe2O3-CS/GCE-1-400 senor. The stability is evaluated by testing the average current response of the modified electrode after prepared 1 day, 1 week, 1 month and 2 month and the biosensor retained over 90% response of its initial sensitivity to the reduction of H2O2, demonstrating its good long-term stability. Cyclic voltammetry experiments were repeatedly performed for 15 times with the G-Fe2O3-CS/GCE-1-400 sensor in the presence of H2O2. The relative standard deviation was approximately 1.89%, which indicated that the reproducibility of as-prepared sensor was excellent.
 | ||
| Fig. 11 Amperometric response of sensor to 5 mM H2O2, AA and UA in 0.1 M pH = 7.4 PBS at −0.38 V. | ||
Conclusion
In summary, a convenient, economic method for preparing a Fe2O3 nanoparticles using SLS as a surfactant by liquid phase-based ultrasonic-assisted method was proposed in this paper. The as-prepared Fe2O3 nanoparticles show a small and homogeneous size distribution with a high specific surface area when the addition amount of SLS in the synthetic system was 1 g, and the α-FeOOH precursor was calcined at 400 °C. The G-Fe2O3-CS/GCE-1-400 modified electrode showed highest electrocatalytic activity toward the detection of H2O2. Chronoamperometry measuring revealed that the prepared senor exhibits high sensitivity, low detection limit, wide linear range, fast response time, good stability and anti-interference ability. Hence, the as-prepared G-Fe2O3-CS/GCE modified electrode provides a effective way for the preparation of the high-performance enzyme-free sensor.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21375050, 61171015), the Open Project Program of State Key Laboratory of Analytical Chemistry for Life Science (Nanjing University) (KLACLS1010), the Program of Department of Education of Jiangsu Province (12KJD610003), and the Natural Science Foundation of Jiangsu Province (BK20131249) for financial support of this research.References
- B. H. Kim, M. J. Hackett, J. Park and T. Hyeon, Chem. Mater., 2014, 26, 59–71 CrossRef CAS.
- X. S. Liu, N. Huang, H. Li, Q. Jin and J. Ji, Langmuir, 2013, 29, 9138–9148 CrossRef CAS PubMed.
- J. S. Son, J. S. Lee, E. V. Shevchenko and D. V. Talapin, J. Phys. Chem. Lett., 2013, 4, 1918–1923 CrossRef CAS.
- Y. D. Yin and D. Talapin, Chem. Soc. Rev., 2013, 42, 2484–2487 RSC.
- N. Iqbal, A. Afzal and A. Mujahid, RSC Adv., 2014, 4, 43121–43130 RSC.
- D. W. Li, L. Luo, Z. Y. Pang, X. D. Chen, Y. B. Cai and Q. F. Wei, RSC Adv., 2014, 4, 3857–3863 RSC.
- A. Sarkar, A. K. Singh, G. G. Khan, D. Sarkar and K. Mandal, RSC Adv., 2014, 4, 55629–55634 RSC.
- J. Lyytinen, M. Berdova, P. Hirvonen, X. W. Liu, S. Franssila, Q. Zhou and J. Koskinen, RSC Adv., 2014, 4, 37320–37328 RSC.
- X. H. Wang, Y. K. Zhang, C. Hao, F. Feng, H. B. Yin and N. C. Si, Ind. Eng. Chem. Res., 2014, 53, 6585–6592 CrossRef CAS.
- H. Y. Zhao, W. Zheng, Z. X. Meng, H. M. Zhou, X. X. Xu, Z. Li and Y. F. Zheng, Biosens. Bioelectron., 2009, 24, 2352–2357 CrossRef CAS PubMed.
- Z. L. Liu, K. X. Wang, L. Xiao, X. J. Chen, X. D. Ren, J. T. Lu and L. Zhuang, RSC Adv., 2014, 4, 37701–37704 RSC.
- S. H. Chen, R. Yuan, Y. Q. Chai, L. G. Zhang, N. Wang and X. L. Li, Biosens. Bioelectron., 2007, 22, 1268–1274 CrossRef CAS PubMed.
- K. F. Zhou, Y. H. Zhu, X. L. Yang, J. Luo, C. Z. Li and S. R. Luan, Electrochim. Acta, 2010, 55, 3055–3060 CrossRef CAS PubMed.
- K. J. Huang, D. J. Niu, X. Liu, Z. W. Wu, Y. Fan, Y. F. Chang and Y. Y. Wu, Electrochim. Acta, 2011, 56, 2947–2953 CrossRef CAS PubMed.
- M. G. Li, S. D. Xu, M. Tang, L. Liu, F. Gao and Y. L. Wang, Electrochim. Acta, 2011, 56, 1144–1149 CrossRef CAS PubMed.
- Z. F. Wang, F. Liao, T. T. Guo, S. W. Yang and C. M. Zeng, J. Electroanal. Chem., 2012, 664, 135–138 CrossRef CAS PubMed.
- T. S. Liu, T. F. Kang, L. P. Lu, Y. Zhang and S. Y. Cheng, J. Electroanal. Chem., 2009, 632, 197–200 CrossRef CAS PubMed.
- S. H. Chen, R. Yuan, Y. Q. Chai, L. Y. Zhang, N. Wang and X. L. Li, Biosens. Bioelectron., 2007, 22, 1268–1274 CrossRef CAS PubMed.
- M. Shamsipura, M. Asgari, M. G. Maragheh and A. A. Moosavi-Movahedi, Bioelectrochemistry, 2012, 83, 31–37 CrossRef PubMed.
- J. Hong, W. Y. Yang, Y. X. Zhao, B. L. Xiao, Y. F. Gao, T. Yang, H. Ghourchian, Z. Moosavi-Movahedid, N. Sheibani, J. G. Li and A. A. Moosavi-Movahedi, Electrochim. Acta, 2013, 89, 317–325 CrossRef CAS PubMed.
- L. Aghebati-maleki, B. Salehi, R. Behfar, H. Saeidmanesh, F. Ahmadian, M. Sarebanhassanabadi and M. Negahdary, Int. J. Electrochem. Sci., 2014, 9, 257–271 Search PubMed.
- M. Shamsipur, M. Asgari, M. F. Mousavi and R. Davarkhah, Electroanalysis, 2012, 24, 357–367 CrossRef CAS.
- A. T. Ezhil Vilian and S. M. Chen, RSC Adv., 2014, 4, 55867–55876 RSC.
- B. B. Jiang, X. W. Wei, F. H. Wu, K. L. Wu, L. Chen, G. Z. Yuan, C. Dong and Y. Ye, Microchim. Acta, 2014, 181, 1463–1470 CrossRef CAS.
- L. M. Li, Z. F. Du, S. Liu, Q. Y. Hao, Y. G. Wang, Q. H. Li and T. H. Wang, Talanta, 2010, 82, 1637–1641 CrossRef CAS PubMed.
- A. P. Liu, W. J. Dong, E. J. Liu, W. H. Tang, J. Q. Zhu and J. C. Han, Electrochim. Acta, 2010, 55, 1971–1977 CrossRef CAS PubMed.
- Z. Y. Miao, D. Zhang and Q. Chen, Materials, 2014, 7, 2945–2955 CrossRef CAS PubMed.
- H. Y. Sun, H. Q. Yuan, Z. M. Liu, B. X. Han and X. R. Zhang, Adv. Mater., 2005, 17, 2993–2997 CrossRef.
- M. Y. Zhu, Y. Wang, D. H. Meng, X. Z. Qin and G. W. Diao, J. Phys. Chem. C, 2012, 116, 16276–16285 CAS.
- S. W. Cao and Y. J. Zhu, J. Phys. Chem. C, 2008, 112, 6253–6257 CAS.
- Q. Zhang, X. W. Lu, L. Y. Chen, Y. X. Shi, T. Xu and M. L. Liu, Mater. Lett., 2013, 106, 447–451 CrossRef CAS PubMed.
- X. M. Liu, S. Y. Fu, H. M. Xiao and C. J. Huang, J. Solid State Chem., 2005, 178, 2798–2803 CrossRef CAS PubMed.
- S. Bai, X. P. Shen, X. Zhong, Y. Liu, G. X. Zhu, X. Xu and K. M. Chen, Carbon, 2012, 50, 2337–2346 CrossRef CAS PubMed.
- X. H. Liao, J. J. Zhu, W. Zhong and H. Y. Chen, Mater. Lett., 2001, 50, 341–346 CrossRef CAS.
- B. Zhao, Y. Wang, H. Guo, J. Wang, Y. He, Z. Jiao and M. Wu, Mater. Sci.-Pol., 2007, 25, 1143–1148 CAS.
- G. Yu, B. Li, H. S. Wang, C. Liu and X. D. Mu, BioResources, 2013, 8, 1055–1063 Search PubMed.
- D. Ji, Z. Y. Luo, M. He, Y. J. Shi and X. L. Gu, Cem. Concr. Res., 2012, 42, 1199–1206 CrossRef CAS PubMed.
- T. T. Miao, Y. R. Guo and Q. J. Pan, J. Nanopart. Res., 2013, 15, 1725–1737 CrossRef.
- S. Allahyari, M. Haghighi, A. Ebadi and S. Hosseinzadeh, Ultrason. Sonochem., 2014, 21, 663–673 CrossRef CAS PubMed.
- A. Abdelwahab and Y.-B. Shim, Sens. Actuators, B, 2014, 201, 51–58 CrossRef CAS PubMed.
- Z. H. Dai, S. Q. Liu, H. X. Ju and H. Y. Chen, Biosens. Bioelectron., 2004, 19, 861–867 CrossRef CAS PubMed.
- X. J. Liu, J. F Liu, Z. Chang, L. Luo, X. D. Lei and X. M. Sun, RSC Adv., 2013, 3, 8489–8494 RSC.
- M. Magroa, D. Baratella, N. Pianca, A. Toninello, S. Grancarab, R. Zboril and F. Vianello, Sens. Actuators, B, 2013, 176, 315–322 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |