DOI:
10.1039/C4RA17201A
(Paper)
RSC Adv., 2015,
5, 28536-28553
Zr(IV) complexes containing salan-type ligands: synthesis, structural characterization and role as catalysts towards the polymerization of ε-caprolactone, rac-lactide, ethylene, homopolymerization and copolymerization of epoxides with CO2†
Received
29th December 2014
, Accepted 9th March 2015
First published on 10th March 2015
Abstract
Zr(IV) complexes containing salan-type diamine bis(phenolato) ligands were synthesized and characterized by various spectroscopic techniques and X-ray crystallography. We observed the formation of dinuclear Zr complexes where each Zr centre adopts a distorted octahedral geometry. The metal centers are bridged with two isopropoxide groups and each metal center has two terminal isopropoxide moieties attached to it. These compounds show notable activities towards the ring-opening polymerization (ROP) of ε-caprolactone (ε-CL) and rac-lactide (rac-LA) in the presence and absence of benzyl alcohol as an external initiator, resulting in very high number average molecular weight (Mn) polymer with controlled molecular weight distributions (MWDs). There is a close proximity between observed molecular weight (Mobsn) and theoretical molecular weight (Mtheon). Analysis of MALDI-TOF and 1H NMR spectra of low Mn oligomers reveals that the isopropoxide groups as well as the OBn groups initiate the ROP. Kinetic studies reveal that the polymerization follows first-order kinetics and is faster in the presence of benzyl alcohol (BnOH). All the complexes were active precatalysts towards the polymerization of ethylene. In addition, all the complexes were found to copolymerize cyclohexene oxide (CHO) and propylene oxide (PO) with CO2, affording the formation of degradable polycarbonate with moderate Mn values and narrow MWDs. Their thermal properties were examined using DSC and TGA analysis. In the case of styrene oxide (SO), the coupled product of CO2 and SO was observed.
Introduction
There are global environmental concerns associated with the depletion of fossil feed stocks. The synthesis of polymeric materials from monomers derived from biomass is of prime focus since the dependence on non-renewable petroleum resources may be reduced. The ROP of lactides and lactones to produce aliphatic polyester has been of growing interest recently and has been extensively researched for the past few decades owing to their biodegradable, biocompatible and permeable properties.1 Poly(lactic acid) (PLA) is a promising material because its feedstock, lactide, is obtained from annually bio renewable resources such as corn, sugar-beet and dairy products and it degrades to yield nontoxic lactic acid and beyond.2 Hence, PLA has become a valuable alternative for non-renewable petroleum derived products.3 It has found enormous applications in biomedical, pharmaceutical, and agricultural industry such as in the production of adsorbable stitches, resorbable medical implants, controlled drug delivery vehicles, ligating clips, bone pins and scaffolds for tissue engineering.4 In addition, PLA is largely applied in multidisciplinary areas such as production of fibers, plastics and coatings.2b,5 In comparison to the traditional polycondensation, ROP is useful in terms of achieving high Mn polymers with narrow MWDs and thus allow us a reliable method for the synthesis of block copolymer and gives us a scope to achieve control over polymer stereochemistry by proper catalyst design.5b A wide range of catalytic systems derived from magnesium,6–10 zinc,6,8,10–14 calcium,9,10,15,16 aluminum,14,17 yttrium,17j,18 the lanthanides,19 tin,20 the group 4 elements,15,21–23 group 5 metals,24 germanium,25 indium,26 and iron27,28 were investigated to initiate the ROP of cyclic esters yielding polymers with high Mn and narrow MWDs. Although Sn(Oct)2 has been used and accepted by the U.S. Food and Drug Administration for the industrial production of PLA, the toxicity and downstream polymer degradation associated with the metal is the primary concern for biomedical applications.4b,29 Therefore, the synthesis of highly active well defined complexes containing biologically benign metals are most important.15,30 Amongst various protocols available, the coordination–insertion mechanism has been extensively used as it has the ability to produce polymer with high Mn with narrow MWDs.4b,4e,31 PLA can be synthesized in solution32,33 as well as in bulk.34 To minimize the side reactions (inter and intrachain transesterification reactions) and for the execution of the living nature of the polymerization, solution polymerization is preferred over bulk, although the rate of solution polymerization is slower than that of bulk polymerization, and thus not applicable for large scale production of PLA.35 By employing sterically bulky ligands like salen or its analogues, one can minimize the polymerization side reactions such as intramolecular and intermolecular transesterification in an appreciable manner and thus control the polymerization process following chain-end control mechanism.11,36 Previously from our group, we reported dinuclear Zr complexes containing salen ((R,R)-(−)-N,N′-bis(3,5-di-t-butylsalicylidene)-1,2-cyclohexanediamine) ligands and their bulk polymerization activity towards cyclic esters and lactides.37 To the best of our knowledge the polymerization results described in this account are the best for the ROP of ε-CL and rac-LA in terms of activity and the control over polymerization using salan (diamine bis(phenolato)) type dinuclear Zr complexes among other reported catalysts containing group 4 metals.21–23
The depletion of oil resources along with environmental and economical concerns such as global warming and high oil prices have influenced the scientific community for the chemical conversion of carbon dioxide (CO2), allowing the use of it as a renewable carbon source. CO2, despite of its high thermodynamic stability is an attractive chemical feedstock because it is nontoxic, nonflammable, sufficiently abundant in nature with high purity, independent from resources like fossil fuels, inexpensive, nonhazardous, and waste product of many chemical processes, including combustion and fermentation.38,39 Recent literature indicates that CO2 has become an alternative of substances like carbon monoxide or phosgene in many processes.38,40 The production of aliphatic polycarbonates by metal catalyzed copolymerization of carbon dioxide and epoxides was first pioneered 40 years ago by Inoue et al. and this was a promising route for the activation and use of CO2 as a renewable C-1 source.39,41 The aliphatic polycarbonates have enormous applications as insulation foams, adhesives, coatings, ceramic binders, elastomers, packaging materials, rigid plastics as well as in the synthesis of engineering thermoplastics and resins, whilst the cyclic carbonates, a side product in the copolymerization reaction of epoxide and CO2 are used as high boiling solvents, electrolytes, fuel additives and as sustainable reagents.42,43 In addition to their biodegradability and biocompatibility, polycarbonates are used in biomedical applications such as drug delivery vesicles, bone screws and scaffolding or suture wire.44 Initially heterogeneous catalysts were employed for the copolymerization reactions.45 Over the last two decades, the catalytic activities for the copolymerization reactions have increased enormously by the use of homogeneous catalysts.39b,42a,46 A wide variety of homogeneous catalysts employing a variety of metal sites, such as Al,42a,42d,47 Zn,48 Cr,49–51 Co39e,52 and Fe53 have been studied for use in the copolymerization of CO2 and epoxides. Recently Nozaki and co-workers investigated for the first time the copolymerization of CHO and CO2 using tetravalent metal (Ti, Zr, Ge and Sn) complexes, supported by the boxdipy ligand (boxdipy = 1,9-bis(2-oxidophenyl)dipyrrinate). The activities and selectivities toward CHO/CO2 copolymerization were noteworthy.54a Later Roux and co-workers reported copolymerization of CHO with CO2 using tridentate N-heterocyclic carbene titanium(IV) complexes.54b Here decent activities and high selectivities were achieved. Very recently Ko and his research group reported benzotriazole phenolate zirconium complexes and oxo-bridged bimetallic group 4 complexes bearing amine-bis(benzotriazole phenolate) derivatives for the copolymerization of CO2 with CHO. Both the catalytic systems were able to copolymerize effectively with good selectivity and high carbonate-linkages.54c,54d We report for the first time the copolymerization of epoxides (CHO and PO) and coupling of SO and CO2 using zirconium(IV) complexes bearing salan type ligands. Our prime focus was to achieve the same activity and selectivity as above reports. We are delighted to disclose that we got comparable results in terms of yields, activities and selectivities. The turnover frequency (TOF) values are noteworthy. Although CHO and PO has been extensively researched, SO is much less explored. The salen,55 along with most recently the salan ligands which contains the metal sites as Cr49a,50,51c,51f,51h and Co52a,52e,52f,52m,52n,56 have been widely studied for epoxides/CO2 copolymerization. Nucleophilic cocatalyst such as tetrabutylammonium bromide (TBAB) is typically required for the copolymerization process exhibiting high activities due to their easily modifiable steric and electronic properties.50d The electronic and steric properties of the ancillary ligand's substituents have great control over the turnover numbers (TONs) and TOFs in this process.39a,57 In general, electron-withdrawing groups, have often led to higher TONs and TOFs upon reducing the electron density on the metal centers.48b,58 Herein, we report first time the copolymerization of epoxides and CO2 using group 4 metal alkoxide complexes bearing salan ligands yielding polymer with moderate Mn value and narrow MWDs. All the complexes were highly active towards the copolymerization of epoxides (CHO and PO) and CO2 resulting in the formation of degradable polycarbonate with high TONs and TOFs. In addition, their thermal properties were investigated by means of differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) techniques. We observed the formation of cyclic styrene carbonate by the coupling of CO2 and SO. The mechanism of ε-CL and rac-LA polymerizations was investigated by both experimental and theoretical methods. In addition, the activities of all the complexes as precatalyst towards the polymerization of ethylene were examined.
Result and discussion
The ligands (L1–L3) were synthesized according to the literature published procedure.59 At room temperature, in an argon filled glove box, solutions of L1–L3 and Zr(OiPr)4·iPrOH in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometric ratio in dry toluene was stirred for 24 h resulting in the formation of complexes (Scheme 1) with high yields and purity. All the Zr complexes (1–3) were characterized using various spectroscopic techniques and their purity was assured by the correct elemental analysis results. The 1H NMR of aliquots removed from the reaction mixture reveals the disappearance of the O–H signal from the ligands, and the appearance of the resonances for the protons of the alkoxides groups, indicating the formation of the desired metal complexes. The 1H and 13C NMR clearly depict signals corresponding to the terminal and bridging alkoxy groups. In case of complex 1, we observed four distinct peaks (1H NMR) in ratios of 2:2:1:1 (ter:ter:bri:bri) corresponding to the terminal and bridging isopropoxides groups in the range between 4.22–4.27 ppm (ter, 2H), 4.28–4.45 ppm (ter, 2H), 4.48–4.71 ppm (bri, 1H) and 4.87–4.93 ppm (bri, 1H). These clearly indicate that the complexes possess four different types of isopropoxide groups. This was further supported by the 13C NMR signals which appear at 70.46 ppm (ter), 71.17 ppm (ter), 72.07 ppm (bri) and 72.44 ppm (bri). Due to the diastereotopic nature of the four benzylic protons, we noticed two doublets in 1H NMR. As evident by the X-ray analysis of all the complexes, we observed that the cyclohexyl ring is in chair conformation, thus increasing the cavity size provided by the ligands. Electrospray ionization mass spectrometry (ESI-MS) spectra disclosed that these compounds have a dinuclear structure.
:2 stoichiometric ratio in dry toluene was stirred for 24 h resulting in the formation of complexes (Scheme 1) with high yields and purity. All the Zr complexes (1–3) were characterized using various spectroscopic techniques and their purity was assured by the correct elemental analysis results. The 1H NMR of aliquots removed from the reaction mixture reveals the disappearance of the O–H signal from the ligands, and the appearance of the resonances for the protons of the alkoxides groups, indicating the formation of the desired metal complexes. The 1H and 13C NMR clearly depict signals corresponding to the terminal and bridging alkoxy groups. In case of complex 1, we observed four distinct peaks (1H NMR) in ratios of 2:2:1:1 (ter:ter:bri:bri) corresponding to the terminal and bridging isopropoxides groups in the range between 4.22–4.27 ppm (ter, 2H), 4.28–4.45 ppm (ter, 2H), 4.48–4.71 ppm (bri, 1H) and 4.87–4.93 ppm (bri, 1H). These clearly indicate that the complexes possess four different types of isopropoxide groups. This was further supported by the 13C NMR signals which appear at 70.46 ppm (ter), 71.17 ppm (ter), 72.07 ppm (bri) and 72.44 ppm (bri). Due to the diastereotopic nature of the four benzylic protons, we noticed two doublets in 1H NMR. As evident by the X-ray analysis of all the complexes, we observed that the cyclohexyl ring is in chair conformation, thus increasing the cavity size provided by the ligands. Electrospray ionization mass spectrometry (ESI-MS) spectra disclosed that these compounds have a dinuclear structure.
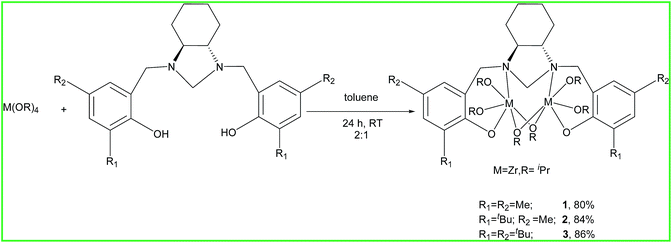 |
| | Scheme 1 Preparation of Zr complexes. | |
Complexes 1–3 were characterized by single crystal X-ray diffraction technique. The studies reveal that these complexes are dinuclear in the solid state where each Zr centre adopts a distorted octahedral geometry (Fig. 1, 2 and Fig. S10, ESI†). The two nitrogen centers from the ligand site are connected to the two Zr centers. Besides, the two Zr centers are bridged with two isopropoxide groups forming a [Zr2O2] four membered ring and each metal center has two terminal isopropoxides moieties attached to it. In addition, the metal centers are at a distances of 3.5361 Å, 3.5508 Å and 3.5480 Å (1–3) respectively. Due to the presence of the cyclohexyl ring in a more thermodynamically stable chair form, the two C–N bonds (cyclohexanediamine) are in a position that it can easily accommodate the two metal centres. Like the Zr(1)–O(1) and Zr(1)–O(2) bond, the Zr(1)–N(1) and Zr(2)–N(2) bond lengths are almost identical. The bridging isopropoxides (O(3)–Zr(2) 2.183(2) vs. O(4)–Zr(2) 1.951(3)) groups have longer bond length than terminal isopropoxides groups. On careful observation of various bond lengths and bond angles, we can conclude that the structure adopts a distorted octahedral geometry (Fig. 1). This dinuclear core is quite similar to the starting material Zr(OiPr)4·iPrOH (Gowda et al., 2009; Cohen et al., 2009).60 The molecular structures for the complexes (1 and 2) are represented below. The molecular structure for 3 is represented in Fig. S10 (ESI†).
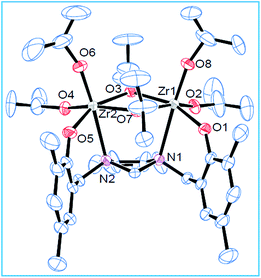 |
| | Fig. 1 Molecular structure of 1; thermal ellipsoids were drawn at 30% probability level. Selected bond lengths (Å) and bond angles (°): O(4)–Zr(2) 1.951(3), O(5)–Zr(2) 2.013(3), O(1)–Zr(1) 2.007(2), O(2)–Zr(1) 1.950(3), O(3)–Zr(2) 2.183(2), N(1)–Zr(1) 2.619(3), N(2)–Zr(2) 2.586(3), O(8)–Zr(1)–O(2) 99.76(13), O(1)–Zr(1)–O(3) 157.32(10), O(8)–Zr(1)–N(1) 174.00(11), O(2)–Zr(1)–Zr(2) 126.36(9), O(8)–Zr(1)–Zr(2) 106.30(9), N(1)–Zr(1)–Zr(2) 76.11(6), O(6)–Zr(2)–O(4) 98.42(13), O(4)–Zr(2)–O(7) 159.41(12). | |
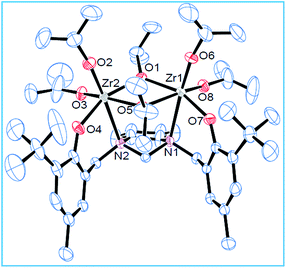 |
| | Fig. 2 Molecular structure of 2; thermal ellipsoids were drawn at 30% probability level. Selected bond lengths (Å) and bond angles (°): O(1)–Zr(2) 2.187(3), O(2)–Zr(2) 1.915(3), O(5)–Zr(2) 2.189(3), O(5)–Zr(1) 2.200(3), O(8)–Zr(1) 1.952(3), N(1)–Zr(1) 2.614(3), O(8)–Zr(1)–O(1) 93.96(12), O(7)–Zr(1)–O(1) 159.21(10), O(8)–Zr(1)–N(1) 84.31(12), O(2)–Zr(2)–Zr(1) 105.52(10), O(3)–Zr(2)–O(4) 97.66(13), N(1)–Zr(1)–Zr(2) 75.70(6), O(6)–Zr(1)–O(8) 100.72(14), O(2)–Zr(2)–O(4) 97.19(13). | |
Polymerization studies
All the complexes proved to be highly active towards the ROP of ε-CL and rac-LA and the observed molecular weights (Mobsn) were found to be in good agreement with the theoretical molecular weights (Mtheon) (Table 1 and Scheme 2). The polymerization results obtained is the fastest so far obtained in group 4 metal catalysts as evident by the time (6 min) and TOF value (33 min−1) for the polymerization process in 200:1 (monomer:catalyst) ratio (Table 1).21–23 The time taken for polymerization was much lesser compared to other group 4 catalytic systems.21–23 Previously from our group, we reported some excellent polymerization results using group 4 metal catalysts but the time of polymerization was higher than this present catalytic system.37 Davidson and his research group reported bulky amine tris(phenolate) complexes as initiator for the ROP of LA.22b But, the time of polymerization was higher and the molecular weight distributions (MWDs) was not narrow enough, indicating less controlled polymerization. Excellent solubility in various solvents and robustness towards decomposition initiated an impetus to study the catalytic behavior of these complexes towards the ROP of ε-CL and rac-LA. From the present results, it is reasonable to conclude that these polymerizations have a great degree of control over the monomer conversion and the Mn value in all the ratios of monomer and catalyst (Tables 1 and 2). The narrow molecular weight distributions indicate well controlled polymerization. A plot of Mn vs. [M]o/[C]o (initial monomer to catalyst ratio) vs. MWDs (Mw/Mn) (Fig. 3), revealed that the variation of Mn with increment in [M]o/[C]o is sharp and also the MWDs remain almost invariable with the variation of ratios for a given monomer. We observed a sharp increment of Mn with % conversion with almost invariable MWDs when we plotted Mn vs. % conversion vs. MWDs (Mw/Mn) (Fig. S11, ESI†). The effect of the ligand substituents in complexes 1–3 on the catalytic activities for ROP of ε-CL and rac-LA was investigated. Complex 1 being least sterically congested showed the highest catalytic activity in terms of Mn value, MWDs and time of polymerization. In this regards, 3 showed lowest activity owing to highly sterically hindered conformation. The reactivity of all the complexes towards the polymerization process was verified by DFT calculations by studying the HOMO–LUMO energy gap of all the complexes. The energy gap in 1 and 3 is 3.18 eV and 5.14 eV respectively (discussed later). The polymerization continues upon the addition of the monomer externally. The polymerizations were performed using 1–3 with rac-LA and ε-CL in the presence of benzyl alcohol (BnOH) in order to examine the effects of external initiator towards the polymerization and the control over polymerization (Table S2, ESI†). The polymerization reactivity is much higher in the presence of BnOH and the general trend of reactivity followed the same order as rac-LA (1 > 2 > 3). A close correlation between the theoretical Mn and observed Mn was found and the MWDs of the resulting polymers were narrow. The molecular weights (Mn) increased linearly with the monomer conversions with almost unchanged MWDs and follow the same trend as found in the absence of alcohol. Homonuclear decoupled 1H NMR spectra shows stereoselectivity with good heterotactic bias (Pr ∼ 0.71) for the isolated polymers (Fig. S12, ESI†).
Table 1 Polymerization data for rac-LA and ε-CL using 1–3 in 200:1 (monomer:catalyst) ratio
| Entry |
Initiator |
Monomer |
T/°C |
Yield (%) |
aTime/min |
bTOF/min |
cMobsn/kg mol−1 |
dMtheon/kg mol−1 |
Mw/Mn |
ePr |
| Time of polymerization measured by quenching the polymerization reaction when all monomer was found consumed. Turnover frequency (TOF) = number of moles of monomer consumed/(mole of catalyst × time of polymerization). Measured by GPC at 27 °C in THF relative to polystyrene standards with Mark–Houwink corrections for Mn. Mn (theoretical) at 100% conversion = [M]o/[C]o × mol wt (monomer) + mol wt (end group).7e Calculated from homonuclear decoupled 1H NMR spectrum.7e |
| 1 |
1 |
rac-LA |
140 |
99 |
6 |
33 |
27.43 |
28.89 |
1.07 |
0.70 |
| 2 |
2 |
rac-LA |
140 |
97 |
15 |
13 |
24.95 |
28.89 |
1.04 |
0.73 |
| 3 |
3 |
rac-LA |
140 |
98 |
55 |
4 |
21.22 |
28.89 |
1.08 |
0.71 |
| 4 |
1 |
ε-CL |
80 |
98 |
22 |
9 |
24.06 |
22.89 |
1.07 |
|
| 5 |
2 |
ε-CL |
80 |
99 |
31 |
6 |
22.88 |
22.89 |
1.10 |
|
| 6 |
3 |
ε-CL |
80 |
98 |
44 |
4 |
21.30 |
22.89 |
1.08 |
|
 |
| | Scheme 2 Polymerization of rac-LA and CL. | |
Table 2 Polymerization data based on changing the ratios in case of rac-LA using 1, 2, and 3 at 140 °C
| Entry |
Initiator |
[M]/[C]ratio |
aTime/min |
bTOF/min |
Yield (%) |
cMobsn/kg mol−1 |
dMtheon/kg mol−1 |
Mw/Mn |
| Time of polymerization measured by quenching the polymerization reaction when all monomer was found consumed. Turnover frequency (TOF) = number of moles of monomer consumed/(mole of catalyst × time of polymerization). Measured by GPC at 27 °C in THF relative to polystyrene standards with Mark–Houwink corrections for Mn. Mn (theoretical) at 100% conversion = [M]o/[C]o × mol wt (monomer) + mol wt (end group).7e |
| 1 |
1 |
100 |
3 |
32 |
96 |
13.90 |
14.47 |
1.05 |
| 2 |
1 |
200 |
6 |
33 |
99 |
27.43 |
28.89 |
1.07 |
| 3 |
1 |
400 |
14 |
28 |
99 |
55.50 |
57.72 |
1.08 |
| 4 |
1 |
600 |
25 |
23 |
97 |
85.39 |
86.54 |
1.10 |
| 5 |
1 |
800 |
39 |
20 |
96 |
113.82 |
115.37 |
1.13 |
| 6 |
1 |
1000 |
101 |
10 |
98 |
143.80 |
144.20 |
1.15 |
| 7 |
2 |
100 |
7 |
14 |
99 |
11.15 |
14.47 |
1.02 |
| 8 |
2 |
200 |
15 |
13 |
95 |
24.95 |
28.89 |
1.04 |
| 9 |
2 |
400 |
31 |
12 |
94 |
51.80 |
57.72 |
1.05 |
| 10 |
2 |
600 |
49 |
12 |
98 |
82.44 |
86.54 |
1.07 |
| 11 |
2 |
800 |
69 |
11 |
98 |
111.58 |
115.37 |
1.08 |
| 12 |
2 |
1000 |
135 |
7 |
99 |
140.3 |
144.20 |
1.10 |
| 13 |
3 |
100 |
30 |
3 |
97 |
9.05 |
14.47 |
1.09 |
| 14 |
3 |
200 |
55 |
3 |
95 |
21.22 |
28.89 |
1.11 |
| 15 |
3 |
400 |
83 |
5 |
98 |
48.2 |
57.72 |
1.12 |
| 16 |
3 |
600 |
105 |
6 |
96 |
78.02 |
86.54 |
1.13 |
| 17 |
3 |
800 |
129 |
6 |
95 |
107.3 |
115.37 |
1.15 |
| 18 |
3 |
1000 |
159 |
6 |
97 |
136.73 |
144.20 |
1.18 |
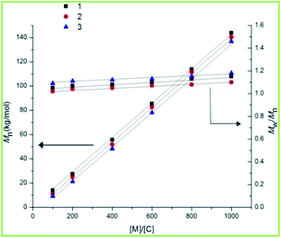 |
| | Fig. 3 Plot of Mn and Mw/Mn vs. [M]o/[C]o for rac-LA polymerization at 140 °C using 1, 2 and 3. | |
Kinetics of polymerization
In the next part of our investigations, we have studied the kinetics of the polymerization for the complex 1, 2 and 3. As the time of polymerization was too less at a ratio of 200:1 we performed kinetic studies for the polymerization of rac-LA in ratio [rac-LA]o/[Zr]o = 600 at 140 °C. A plot of % conversion of rac-LA against time depicts a sigmoidal curve (Fig. S13, ESI†). From the kinetic experiment results, it was ascertained that there is a first-order dependence of the rate of polymerization on rac-LA concentration and no induction period was observed. The plot of ln([rac-LA]o/[rac-LA]t) vs. time was found to be linear (Fig. 4). The values of the apparent rate constant (kapp) for rac-LA polymerization catalyzed by 1, 2 and 3 were evaluated from the slope of these straight lines and were found to be 13.89 × 10−2 min−1, 6.17 × 10−2 min−1 and 2.99 × 10−2 min−1 respectively. From the rate constants of polymerization, it can be concluded that the rate is fastest for 1 followed by 2 and then 3. This is justified by the time taken for the polymerization. For ε-CL also we studied the kinetic experiment using the complex 1, 2 and 3 at 200:1 ratio at a temperature of 80 °C. Here again the results followed the same trend (Fig. S14 and S15, ESI†). The values of the apparent rate constant (kapp) for ε-CL polymerization catalyzed by 1, 2 and 3 were evaluated from the slope of these straight lines and were found to be 16.49 × 10−2 min−1, 9.42 × 10−2 min−1 and 7.23 × 10−2 min−1 respectively. From the rate constants of polymerization, it can be concluded that the rate is fastest for 1 followed by 2 and then 3. This was again justified by the time taken for the polymerization. Later, we studied the kinetic experiments of rac-LA and ε-CL polymerization in the presence of BnOH in 200:1:5 ratio ([M]o/[Cat]o/[BnOH] = 200:1:5) using the complexes 1, 2 and 3. A plot of % conversion of rac-LA and ε-CL against time depicts a sigmoidal curve (Fig. S16 and S17, ESI†). From the linear plot of ln([M]o/[M]t) vs. time for rac-LA (Fig. S18, ESI†) and ε-CL (Fig. S19, ESI†), it was confirmed that the polymerization proceed through first-order kinetics. The values of the apparent rate constant (kapp) for rac-LA polymerization using catalysts 1, 2 and 3 were found to be 27.94 × 10−2 min−1, 14.23 × 10−2 min−1 and 8.93 × 10−2 min−1 respectively, thus the rate is fastest for 1 followed by 2 and then 3. This is justified by the time taken for the polymerization. For ε-CL, the values of the apparent rate constant (kapp) were found to be 15.28 × 10−2 min−1, 12.44 × 10−2 min−1 and 9.63 × 10−2 min−1 respectively, thus following the same trend as above.
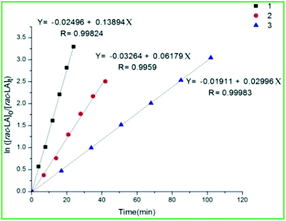 |
| | Fig. 4 Semi-logarithmic plots of rac-LA conversion in time initiated by 1, 2 and 3: [rac-LA]o/[Cat]o = 600 at 140 °C. | |
Mechanism of polymerization
Low molecular weight oligomer of rac-LA and ε-CL were prepared by stirring rac-LA and ε-CL with 1 in 15:1 molar ratio under solvent free conditions at 140 °C and 80 °C respectively. The product was precipitated in cold methanol. The filtered residue was thoroughly dried and examined thoroughly using MALDI-TOF and 1H NMR techniques. In this case, the attached isopropoxides moiety with zirconium in 1 was found to initiate the polymerization. The polymer chain was end capped with isopropoxides ester and hydroxyl group in the both end as ascertained through the analysis of MALDI-TOF and 1H NMR spectra (Fig. S20 and S21, ESI†). In addition, from the MALDI-TOF and 1H NMR spectra analysis it can be implied that the polymerization proceeds through the conventional coordination–insertion mechanism (Scheme S1, ESI†). The polymerization proceeds via the coordination with metal followed by cleavage of acyl oxygen bond of the lactide monomer. As a consequence, the ring opens and the polymer chain starts growing. We observed OBn group as one of the end terminal group when we added benzyl alcohol (15:1:2) as an external alcoholic initiator during the polymerization. This was verified by the end group analysis by MALDI-TOF (Fig. S22, ESI†) and 1H NMR (Fig. S23, ESI†) spectra. In addition, the spectrum reflected the presence of products as a result of intramolecular transesterification product (Fig. S22, ESI†). These can't be differentiated in the 1H NMR (Fig. S23, ESI†). The mechanism for the concerned polymerization is shown in Scheme S1 (ESI†). With ε-CL, a similar procedure was followed and identical conclusions were obtained. These were ascertained using MALDI-TOF (Fig. S24 and S26, ESI†) and 1H NMR (Fig. S25 and S27, ESI†) spectral analysis. The mechanism is shown in Scheme S2 (ESI†).
Density functional theory (DFT) calculations
In order to understand clearly the reactivity of all the complexes towards the ROP, theoretical calculations were carried out at the DFT level using the hybrid functional B3LYP with the Gaussian 09 (ref. 61) suite of programs with LANL2DZ basis set.62 The theoretical results obtained were in good agreement with the experimental results. MPA (Mulliken population analysis) method was carried out on complexes 1 and 3 with the basis set LANL2DZ.62 Mulliken net charges on O-atom of various OiPr groups as well as OPh of ligands in complexes 1 and 3 are summarized in Table S3 (ESI†). From this, we can conclude that the O-atom on OiPr groups is significantly more electron rich than the oxygen atoms in the ligands i.e., the isopropoxides group is the nucleophilic center (Fig. S28, ESI†). Hence, the isopropoxides group initiates the ROP via coordination–insertion mechanism. In addition, the influence of isopropoxides moiety as initiator for the ring-opening polymerization of rac-LA and ε-CL by Zr complexes as catalyst was also studied using DFT calculations.24a,63 The calculated HOMO of the complexes 1 distinctly shows that the electron density is located mainly on the isopropoxides moiety i.e., the isopropoxides group is the nucleophilic center (Fig. 5). Hence, the isopropoxides group initiates the ring opening polymerization via coordination–insertion mechanism. The ligand has practically no influence in ROP. The mechanism of polymerization follows the path of carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) insertion into the metal–O bond of the isopropoxides groups resulting in the formation of polymer that has an isopropoxides end group. For all solid state structures HOMO (highest occupied molecular orbitals) and LUMO (lowest unoccupied molecular orbitals) energy gaps are calculated at the B3LYP/LANL2DZ level (Fig. 6). HOMO and LUMO energy gap explains the eventual absolute hardness and the stability of the complexes. With the increase in the HOMO and LUMO energy gap, the hardness increases and the stability of the complexes also increase.64 The low energy difference between HOMO and LUMO for all the complexes results in high reactivity towards ROP.65 This explains the reactivity order (1 > 2 > 3).
O) insertion into the metal–O bond of the isopropoxides groups resulting in the formation of polymer that has an isopropoxides end group. For all solid state structures HOMO (highest occupied molecular orbitals) and LUMO (lowest unoccupied molecular orbitals) energy gaps are calculated at the B3LYP/LANL2DZ level (Fig. 6). HOMO and LUMO energy gap explains the eventual absolute hardness and the stability of the complexes. With the increase in the HOMO and LUMO energy gap, the hardness increases and the stability of the complexes also increase.64 The low energy difference between HOMO and LUMO for all the complexes results in high reactivity towards ROP.65 This explains the reactivity order (1 > 2 > 3).
 |
| | Fig. 5 DFT calculations (B3LYP, LANL2DZ) showing HOMO of 1 representing electron density on isopropoxide groups. | |
 |
| | Fig. 6 Frontier molecular orbital diagrams of complexes 1 and 3 associated with HOMO–LUMO energy gap of 3.18 eV and 5.14 eV respectively. | |
Copolymerization of epoxides (CHO and PO) and CO2
After having obtained good results for the ROP of rac-LA and ε-CL, we were curious to see the activity of all the complexes towards the copolymerization of epoxides (CHO and PO) and CO2 (Scheme 3). Interestingly, we got excellent results employing group IV metal-alkoxides as potent initiators for such reactions (Table 3). Either in the absence of CO2 or in the absence of cocatalyst such as PPN chloride/azide [PPN = bis(triphenylphosphine)iminium], tetrabutylammonium bromide (TBAB), dimethylaminopyridine (DMAP) or N-methylimidazole (N-MeIm), all the complexes were active catalysts for the homopolymerization of neat CHO, SO and PO yielding a highly viscous liquid of polycyclohexene oxide (PCHO), polystyrene oxide (PSO) and polypropylene oxide (PPO) respectively (Fig. S29–S31, ESI†). We were interested in making the biodegradable copolymer of epoxides and CO2 with a great deal of control over the polymerization process. The unwanted side reaction in this copolymerization studies is the homopolymerization of the epoxides, generating polyether since the reactivity of the epoxides is much higher than that of CO2. A suitable copolymerization catalyst should not result in the homopolymerization product to a great extent. In the absence of cocatalyst, due to the presence of lewis acidic metal centers like Zr(IV), homopolymerization product is most desirable. In the presence of cocatalyst, the Lewis acidity of the metal centers alters significantly.47a In addition, the cocatalyst can act as an additional initiator which can start extra polymer chain.50c That's why we got predominantly copolymerization product. As PPN chloride and PPN azide are not soluble in the monomers involved we carried out the copolymerization reaction using TBAB as a cocatalyst for the copolymerization reactions. In the presence of cocatalysts, it follows a bimolecular mechanism, involving both the catalyst and a cocatalyst.52h,66 It was observed by both Nakano et al. and Luinstra et al. that to facilitate CO2 insertion reactions and prevent decarboxylation, the bidentate coordination mode of the carbonate growing polymer chain is most favorable.51f,67
 |
| | Scheme 3 Copolymerization of CHO and PO with CO2 using TBAB as cocatalyst. | |
Table 3 Polymerization data for the copolymerization of epoxides (CHO and PO) and CO2 using 1, 2 and 3g
| Entry |
I |
M |
M:I:CC |
Time (h) |
T (°C) |
% yielda |
% carbonateb |
%PCc |
TONd |
TOF (h)e |
Mnf kg mol−1 |
Mw/Mnf |
| Determined on the basis of 1H NMR spectroscopy of the crude product obtained. % carbonate = percentage selectivity for carbonate linkages (PCHC + CHC), as determined from the normalized integrals in the 1H NMR spectra using the methylene resonances, including PCHC (δ 4.73 ppm), copolymer ether linkages (δ 3.48 ppm), and CHC (δ 4.01 ppm). Selectivity for PCHC within carbonate products. Turnover number (TON) = number of moles of epoxide consumed per mole of catalyst. Turnover frequency (TOF) = TON/h. Determined by GPC, in THF, using narrow Mn polystyrene standards, for calibration. I = initiator, M = monomer, CC = cocatalyst, T = temperature, PC = polycarbonate, TON = turnover number, TOF = turnover frequency. Applied pressure = 35 bar. |
| 1 |
1 |
CHO |
1000:1:1 |
6 |
50 |
97 |
95 |
98 |
950 |
158 |
16.02 |
1.09 |
| 2 |
1 |
CHO |
1000:1:0.5 |
6 |
50 |
96 |
94 |
97 |
940 |
157 |
15.89 |
1.11 |
| 3 |
1 |
CHO |
1000:1:5 |
6 |
50 |
82 |
81 |
80 |
810 |
135 |
12.12 |
1.23 |
| 4 |
1 |
CHO |
1000:2:1 |
6 |
50 |
77 |
79 |
82 |
790 |
132 |
12.03 |
1.32 |
| 5 |
2 |
CHO |
1000:1:1 |
6 |
50 |
96 |
93 |
95 |
930 |
155 |
14.40 |
1.13 |
| 6 |
3 |
CHO |
1000:1:1 |
6 |
50 |
95 |
90 |
92 |
900 |
150 |
13.39 |
1.16 |
| 7 |
1 |
PO |
1000:1:1 |
6 |
40 |
91 |
80 |
81 |
800 |
133 |
12.56 |
1.07 |
| 8 |
1 |
PO |
1000:1:0.5 |
6 |
40 |
89 |
78 |
80 |
780 |
130 |
12.20 |
1.10 |
| 9 |
1 |
PO |
1000:1:5 |
6 |
40 |
83 |
67 |
65 |
670 |
112 |
10.89 |
1.21 |
| 10 |
1 |
PO |
1000:2:1 |
6 |
40 |
80 |
65 |
68 |
650 |
108 |
10.80 |
1.33 |
| 11 |
2 |
PO |
1000:1:1 |
6 |
40 |
88 |
76 |
79 |
760 |
127 |
12.38 |
1.09 |
| 12 |
3 |
PO |
1000:1:1 |
6 |
40 |
85 |
73 |
76 |
730 |
122 |
11.98 |
1.10 |
The polymerizations are well controlled, with the polymer molecular weight (Mn) increasing linearly with monomer (CHO and PO) conversion and yielding polymers with narrow MWDs (≤1.2) (Fig. 7). The end groups analysis by the MALDI-TOF spectra of the copolymer synthesized from CHO catalyzed by 1 showed that the polymer had hydroxyl groups as end terminals, indicating a chain transfer reactions (Fig. S32, ESI†).47c,52i In addition MALDI-TOF spectra showed the bimodal distributions.53,55c The copolymers were also analyzed by 1H NMR spectroscopy, where the protons adjacent to the carbonate linkage resonated at 4.73 ppm, while the presence of a small peak at 3.48 ppm showed there were little polyether linkages (Fig. 8). 13C NMR assignments of the resulting poly(cyclohexene carbonate) (PCHC) revealed a random distribution of isotactic and syndiotactic carbonate, thus all the polymers were atactic (Fig. 9).68 In case of poly(propylene carbonate) (PPC) we observe methylene (–CH2–) and methine (OCH(Me)–) signals at 3.88–3.92 ppm and 4.72–4.77 ppm respectively in the 1H NMR spectrum (Fig. S33, ESI†). The small peak at 4.44 ppm indicates the presence of little cyclic propylene carbonate product. In 13C NMR spectroscopy methylene (–CH2–) and methine (OCH(Me)–) signals observed at 70.54 ppm and 73.59 ppm respectively. The carbonyl carbon signal of the polycarbonate containing product appeared at 155.06 ppm (Fig. S34, ESI†).
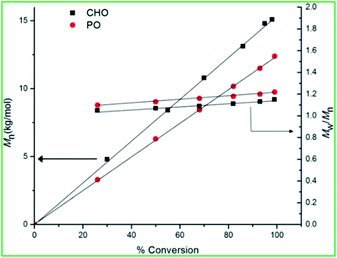 |
| | Fig. 7 Plot of Mn and Mw/Mn vs. % conversion for CHO and PO copolymerization at 50 °C and 40 °C respectively using 1. | |
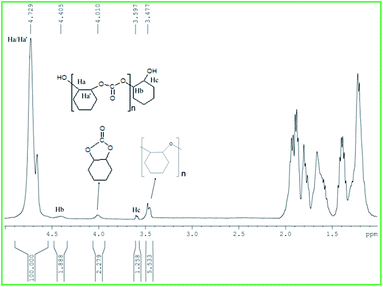 |
| | Fig. 8 1H NMR spectrum of copolymer (PCHC) produced in CDCl3. Peak at (δ = 4.73 ppm) is assigned to the carbonate peak, peaks at (4.40 and 3.6 ppm) are assigned to the methine groups on the end group, OCHC4H8CHOH (Hb) and OCHC4H8CHOH (Hc) respectively. Peak at 4.01 is assigned to the trans-cyclic carbonate. The peak at 3.48 ppm confirms the presence of ether linkages. | |
 |
| | Fig. 9 13C NMR spectrum of poly(cyclohexene carbonate) in the carbonate region produced from cyclohexene oxide and CO2. | |
The time and temperature for the copolymerization reactions were also optimized and with time the reaction rate decreases due to an increase in the viscosity of the reaction mixture. With time, Mn decreases for all the complexes due to the initial homopolymerization generating high molecular weight poly(cyclohexene oxide) and poly(propylene oxide) chains (maximum percentage of the total polymer chains) followed by copolymerization and chain transfer reactions to provide shorter chains. At higher temperatures, thermodynamically favorable cyclic carbonate is formed in an unwanted side reaction.69 In our experiments, with varying time and temperature, the formation of cyclic carbonate product did not exceed 5%. At 50 °C and with a CO2 pressure of 35 bar we got predominantly PCHC with very less polyether linkages (∼5%) and even smaller component of CHC (∼2%) from the copolymerization of CHO and CO2 using catalyst 1 (Fig. 8). The TOF value of 158 and the selectivity over PCHC was noteworthy. The variation of cocatalyst amount from 1 equivalent (Table 3, entry 1) to 0.5 equivalents (Table 3, entry 2), did not have a significant effect on the activity. We got lower Mn as well as lower selectivity using a higher loading of cocatalyst (Table 3, entry 3). The catalyst loading also found to have significant effect on copolymerization reaction. Using a 0.1% catalyst loading vs. epoxide monomer (Table 3, entry 1) we observed better selectivity for polycarbonate, high yield and higher Mn. Lower Mn with broader MWDs was obtained when we used a 0.2% catalyst loading with cocatalyst (Table 3, entry 4). The copolymerization of propylene oxide (PO) with CO2 was carried out at 40 °C for 6 h by using the complexes 1–3 as catalysts. In the absence of any additive, the copolymerization did not proceed and only polypropylene oxide (PPO) was formed. In contrast, the addition of TBAB resulted in the successful production of PPC with a TOF of 151 and moderate PPC selectivity (PPC/PC = 81/19) (Table 3, entry 7). With the increase in temperature, PPC selectivity decrease as the probability of formation of polypropylene oxide (PPO) from the homopolymerization of PO increases. The PPC selectivity was almost independent to the molar ratio of 1–3/TBAB (entries 7 and 8). The PPC selectivity was also decreased when a larger excess amount of TBAB was used (entry 9).
In the case of CHO, we got less yield of the cyclic carbonate product than that of PO because of the steric bulk of the chain formed from CHO was thought to deter the back biting required for cyclic carbonate formation as shown in Scheme 2. In spite of being more costly with respect to PO, CHO is popularly used as the tendency of CHO to form copolymer over cyclic product has made it the commonest monomer for the copolymerization of epoxides with CO2 in recent literature concerning polycarbonate synthesis.46–58 Another advantage of PCHC is its relatively high glass transition temperature (Tg) (114 °C) compared with PPC (42 °C). PPC having lower glass transition temperature of 42 °C, limits its versatile utility. A higher Tg than 40 °C is necessary to allow its use as a stiff plastic material and a lower Tg than room temperature is essential for the use as a soft film.52p In addition, the low decomposition temperature of ∼180 °C, prevents blending with other polymers which can be processed above 200 °C.
The thermal properties of the copolymers synthesized from CHO and PO were also investigated by TGA and DSC measurements. TGA analyses of the copolymer samples indicate that PCHC is thermally stable up to ∼84 °C, with 5%, 50% and 95% degradation occurring by ∼104 °C, 164 °C and 612 °C respectively (Table 4, entry 1). In case of PPC, the temperature for 5%, 50% and 95% weight loss are 64 °C, 122 °C and 291 °C respectively (Table 4, entry 4). The decomposition temperature (Td) values obtained for the copolymer synthesized was less than the previously reported protocol.51e,52p,70 This can be explained by the fact that the copolymers have substantial amount of ether linkages as well as the polymer's molecular weight has a significant role in Td determination. A representative TGA trace and derivative plot is shown in Fig. S35 and S36 (ESI†).
Table 4 DSC and TGA measurements for the different copolymers obtained in Table 3
| Entry |
Initiator |
Copolymers |
Tga (°C) |
Td5b (°C) |
Td50b (°C) |
Td95b (°C) |
| Tg values represent the midpoint temperature during the second heating cycle determined by DSC. Td5, Td50, and Td95 are the decomposition temperature at 5%, 50%, and 95% weight loss, respectively determined by TGA analysis. |
| 1 |
1 |
PCHC |
61 |
104 |
164 |
612 |
| 2 |
2 |
PCHC |
58 |
97 |
155 |
598 |
| 3 |
3 |
PCHC |
56 |
92 |
151 |
595 |
| 4 |
1 |
PPC |
35 |
64 |
122 |
291 |
| 5 |
2 |
PPC |
33 |
62 |
119 |
287 |
| 6 |
3 |
PPC |
30 |
59 |
115 |
285 |
DSC measurements of the PCHC and PPC afforded Tg ranging from 56 °C to 61 °C and 30 °C to 35 °C respectively. The DSC trace and observable Tg for PCHC and PPC produced by complex 1, Table 4, entry 1 and 4 are given in Fig. S37 and S38 (ESI†). The Tg values obtained are found to be less than the values reported in the literature.52p,71 The reason may be the copolymers have small amount of ether linkages. The differences in Tg for PCHC between entries 1, 2 and 3 (61 °C vs. 58 °C vs. 56 °C) is attributed to the variation of molecular weight for the three complexes (Table 4).
Coupling of SO and CO2
Next, we performed the reactions of SO and CO2 (Scheme 4). All the reactions were conducted in neat SO and initiated by complexes 1–3. In all cases, the cyclic product, styrene carbonate (SC) was obtained as the only product. We didn't get any copolymerization product. The 1H NMR spectra of the products were in very good agreement with the previously characterized SC (Fig. 10).72 The carbonyl region peak at 154.73 ppm in the 13C NMR spectrum of styrene carbonate (SC) confirms the coupling of SO and CO2 (Fig. S39, ESI†). All the complexes 1–3 showed notable activity for the formation of styrene carbonate at a temperature of 80 °C and CO2 pressure of 35 bar using TBAB as cocatalyst (Table 5). At room temperature using TBAB as cocatalyst, and maximum CO2 pressure of 55 bar did not yield any carbonate containing product (Table 5, entry 2). The % conversion, TON and TOF values were found noteworthy (Table 5). The production of poly(styrene carbonate) by the copolymerization of SO/CO2 is very difficult due to the occurrence of the ring opening at the methine Cα–O bond rather than the methylene Cβ–O bond (Scheme 5).72,73 That's the reason, the formation of styrene carbonate by intramolecularly cyclic elimination can't be avoided. The electron withdrawing nature of the aromatic ring in styrene oxide causes backbiting of the propagating polycarbonate anion to the benzyl carbon of the adjacent carbonate unit (Scheme 6).72 Some reports with Co(salen) catalysts using suitable cocatalyst has produced alternating poly(styrene carbonate) with an increase selectivity and thermal stability.72,74,75
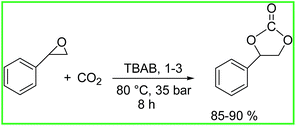 |
| | Scheme 4 Coupling of SO and CO2 to give styrene carbonate (SC). | |
 |
| | Fig. 10 1H NMR spectrum of an aliquot from the reaction mixture of SO/CO2 in CDCl3 (Table 5, entry 1). | |
Table 5 Coupling reaction of SO and CO2 catalyzed by 1, 2 and 3c
| Entry |
I |
T (°C) |
Pressure (bar) |
% yield |
TONa |
TOFb (h) |
| Turnover number (TON) = number of moles of epoxide consumed per mole of catalyst. Turnover frequency (TOF) = TON/h. M:I:CC = 1000:1:1. I = initiator, M = monomer, CC = cocatalyst, T = temperature, TON = turnover number, TOF = turnover frequency. Time = 8 h. |
| 1 |
1 |
80 |
35 |
90 |
900 |
113 |
| 2 |
1 |
25 |
55 |
— |
— |
— |
| 3 |
2 |
80 |
35 |
88 |
880 |
110 |
| 4 |
3 |
80 |
35 |
85 |
850 |
106 |
 |
| | Scheme 5 Difference in nucleophilic ring-opening between styrene oxide and propylene oxide. | |
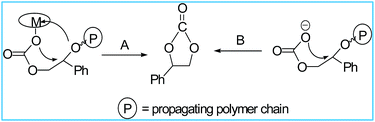 |
| | Scheme 6 Formation of styrene carbonate by intramolecular cyclic elimination of the propagating polymer chains involving the carbonate unit associated with ring-opening of styrene oxide at the methine Cα–O bond. | |
Ring opening homopolymerization of epoxides
Later, we explored the catalytic activity of all the complexes towards the homopolymerization of CHO, PO and SO. The results are summarized in Table 6. All the polymerization reactions of CHO, PO and SO were performed at 80 °C, 30 °C and 120 °C respectively employing two different monomer to catalyst ratios of 1000:1 and 10000:1 respectively under solvent free condition (Scheme 7). The molecular weight and yield of poly(cyclohexene oxide) (PCHO), poly(propylene oxide) (PPO) and poly(styrene oxide) (PSO) increases with the increase in reaction time. The conversion of all the polymers can be measured using 1H NMR spectroscopy by comparing the polymer and unreacted monomer peaks (Fig. S29–S31†). The reactivity of all the complexes shows the same pattern like the ROP of lactide or the copolymerization of epoxides and CO2. CHO appeared to be most reactive as compared to PO and SO towards the homopolymerization reactions.
Table 6 Ring opening homopolymerization of epoxides using 1–3e
| Entry |
I |
M |
M:I |
Time (h) |
T (°C) |
% yielda |
TONb |
TOFc (h) |
Mnd kg mol−1 |
Mw/Mnd |
| Determined on the basis of 1H NMR spectroscopy of the crude product obtained. Turnover number (TON) = number of moles of epoxide consumed per mole of catalyst. Turnover frequency (TOF) = TON/h. Determined by GPC, in THF, using narrow Mn polystyrene standards, for calibration. I = initiator, M = monomer, T = temperature, TON = turnover number, TOF = turnover frequency. |
| 1 |
1 |
CHO |
1000:1 |
6 |
80 |
96 |
960 |
160 |
93.02 |
1.18 |
| 2 |
2 |
CHO |
1000:1 |
6 |
80 |
94 |
940 |
157 |
91.40 |
1.20 |
| 3 |
3 |
CHO |
1000:1 |
6 |
80 |
93 |
930 |
155 |
90.89 |
1.25 |
| 4 |
1 |
CHO |
10000:1 |
48 |
80 |
83 |
8300 |
173 |
901.03 |
1.25 |
| 5 |
2 |
CHO |
10000:1 |
48 |
80 |
79 |
7900 |
165 |
882.12 |
1.28 |
| 6 |
3 |
CHO |
10000:1 |
48 |
80 |
77 |
7700 |
160 |
870.39 |
1.33 |
| 7 |
1 |
PO |
1000:1 |
8 |
30 |
93 |
940 |
118 |
52.78 |
1.20 |
| 8 |
2 |
PO |
1000:1 |
8 |
30 |
92 |
920 |
115 |
49.98 |
1.23 |
| 9 |
3 |
PO |
1000:1 |
8 |
30 |
89 |
890 |
111 |
47.67 |
1.29 |
| 10 |
1 |
PO |
10000:1 |
52 |
30 |
80 |
8000 |
154 |
498.12 |
1.31 |
| 11 |
2 |
PO |
10000:1 |
52 |
30 |
77 |
7700 |
148 |
462.39 |
1.35 |
| 12 |
3 |
PO |
10000:1 |
52 |
30 |
75 |
7500 |
144 |
440.03 |
1.37 |
| 13 |
1 |
SO |
1000:1 |
12 |
120 |
92 |
920 |
77 |
91.40 |
1.22 |
| 14 |
2 |
SO |
1000:1 |
12 |
120 |
90 |
900 |
75 |
86.02 |
1.29 |
| 15 |
3 |
SO |
1000:1 |
12 |
120 |
88 |
880 |
73 |
88.89 |
1.27 |
| 16 |
1 |
SO |
10000:1 |
56 |
120 |
78 |
7800 |
139 |
940.23 |
1.32 |
| 17 |
2 |
SO |
10000:1 |
56 |
120 |
75 |
7500 |
134 |
910.34 |
1.35 |
| 18 |
3 |
SO |
10000:1 |
56 |
120 |
72 |
7200 |
129 |
895.98 |
1.39 |
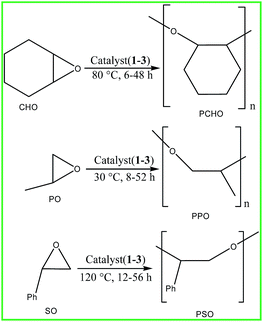 |
| | Scheme 7 Homopolymerization of CHO, PO and SO. | |
Ethylene polymerization
In the next segment of this work, we wanted to study the catalytic activity of all the complexes as precatalyst towards the polymerization of ethylene. Complexes 1–3 were found to be viable initiators towards the polymerization of ethylene. The catalytic activity of all the complexes was examined in the presence of MAO as a cocatalyst. The polymerizations were performed at 50 °C in hexane. From Table 7, it can be concluded that the Mn and activity are greater for complex 1 followed by 2 and 3. These polymerizations were done in various solvents to investigate the effect on activity of these catalysts. From Fig. 11, it may be inferred that the catalytic activity of 1 is maximum in the presence of least polar hexane and minimum in most polar CHCl3. The reason behind this is CHCl3 being most polar, has a stronger solvation effect on the monomer than that of least polar hexane, which prevented the monomer from attacking the cationic species, and propagation for the polymerization is hindered.5 The variation of the activity with various [MAO]/[C] ratios from 500 to 4000 for 1, 2 and 3 revealed that [MAO]/[C] ratio 1000 is optimum for performing these polymerizations as other ratios led to the reduction in the activity (Fig. S40, ESI†). The polymerization proceeds through the activation of the catalyst using MAO as a result of abstraction of an alkoxide moiety and proceeds through a cationic intermediate, which is the true active species. Such abstraction of an alkoxide group by MAO is well established in the literature.76 The reactivity of these catalysts is not exceptional. In most cases, our activities are lower than Fujita's catalysts.77
Table 7 Polymerization data for ethylene using 1–3 along with MAOd
| Entry |
Ia |
Ab |
Yieldc (g) |
Mw kg mol−1 |
Mn kg mol−1 |
Mw/Mn |
| All experiments were performed in hexane at MAO: initiator ratio = 1000, ethylene pressure = 8 atm, 50 °C for 30 min, catalyst = 50 mg, solvent = 45 mL. A = activity in (g PE per mol cat × h) × 104. g of PE obtained after 30 min. I = initiator. |
| 1 |
1 |
6.88 |
1.85 |
99.45 |
55.56 |
1.79 |
| 2 |
2 |
6.49 |
1.60 |
102.70 |
53.21 |
1.93 |
| 3 |
3 |
6.24 |
1.42 |
103.92 |
52.22 |
1.99 |
 |
| | Fig. 11 Catalytic activity of 1 in different solvents for ethylene polymerization. | |
Conclusion
We have synthesized new Zr alkoxide complexes containing salan-type diamine bis(phenolato) ligands. These compounds are potent initiators towards the polymerization of ε-CL and rac-LA and are precatalysts for ethylene polymerization. We have achieved high degree of control over Mn and MWDs. In case of rac-LA and ε-CL polymerizations, there was a very close correlation between observed molecular weight and theoretical molecular weight and the polymerization proceeds in a controlled fashion. The complex 1 is the best catalyst amongst the three complexes as evident by the TOF values (33 vs. 13 vs. 4). The isopropoxide group is responsible for initiating the ROP instead of the ligands, as understood from the MALDI-TOF and 1H NMR spectral analysis and DFT calculations. In the presence of BnOH, the polymers contain the OBn moiety as one of the end groups. Homonuclear decoupled spectra of the PLA formed showed that the polymer was heterotactically enriched. Tacticity is not influenced in an appreciable manner with increasing steric bulk of the complexes. In addition, all the complexes have shown to viable activity for the copolymerization of CO2 and epoxides (CHO and PO), yielding polymers with moderate molecular weights and narrow MWDs. The thermal properties of the copolymer synthesized were analyzed by DSC and TGA analyses. In case of SO, we got cyclic styrene carbonate as the only product. Using MAO as a cocatalyst, we got good results in the ethylene polymerizations.
Experimental section
All reactions were done under a dry argon atmosphere using standard Schlenk techniques or using glove box techniques with the rigorous exclusion of moisture and air. Toluene was dried by heating under reflux for 6 h over sodium and benzophenone and distilled fresh prior to use. Aqueous formaldehyde (30–38%), 2,4-di-tert-butylphenol (99%), 2,4-dimethylphenol (97%), 2-tert-butyl,4-methylphenol (99%) were purchased from Aldrich and used without further purification. Trans-1,2-Diaminocyclohexane (99%) was purchased from Aldrich dried over CaH2 overnight and distilled fresh prior to use. CDCl3 used for NMR spectral measurements was dried over calcium hydride for 48 h, distilled and stored in a glove box. Zr(OiPr)4·iPrOH, was purchased from Sigma-Aldrich and used without further purification. The rac-LA and ε-CL were purchased from Sigma-Aldrich. The rac-LA was purified by sublimation repeatedly and stored in a glove box. The ε-CL was purified by drying over calcium hydride overnight and distilled fresh prior to use. CHO, PO and SO were purchased from Sigma-Aldrich, dry over CaH2 for 24 h and distilled fresh under nitrogen atmosphere prior to use and stored in a glove box. TBAB was purchased from Aldrich and recrystallized in acetone for further purification. MAO was purchased from Aldrich and used without further purification. Research grade CO2 gas for the copolymerization studies and high pure ethylene gas were purchased from indogas, Bangalore, India.
1H and 13C NMR spectra were recorded with a Bruker Avance 400 or Bruker Ascend 500 instrument. Chemical shifts for 1H and 13C NMR spectra were referenced to residual solvent resonances and are reported as parts per million relative to SiMe4. ESI-MS spectra of the samples were recorded using Waters Q-Tof micro mass spectrometer. Elemental analyses were performed with a Perkin Elmer Series 11 analyzer. MALDI-TOF measurements for PLA and poly(caprolactone) (PCL) were done on a Bruker Daltonics or Bruker Ultraflextreme instrument in dihydroxy benzoic acid (DHBA) matrix. For the analysis of poly(cyclohexene carbonate) (PCHC), dithranol matrix in THF at a loading of 1:5 with NaOAc as the cationizing agent was used. The ligands, L1–L3 were prepared according to the literature reported procedures.59
Synthesis and characterization
A general procedure for the synthesis of 1–3 is outlined below: In an argon filled glove box, to a stirred solution of Zr(OiPr)4·iPrOH (0.14 mmol) in 5 mL of dry toluene at −24 °C was added a solution of L1–L3 (0.28 mmol) in 5 mL of dry toluene. The reaction mixture was allowed to warm to room temperature and stirred for additional 24 h. The solvent was removed under reduced pressure and the white residue obtained was crystallized from concentrated toluene solution at −24 °C.
Compound 1. Yield 0.06 g, (80%). Mp: 143 °C. 1H NMR (400 MHz, CDCl3): δ = 0.63–0.65 (d, J = 8 Hz, CH(CH3)2, 2H), 0.99–1.01 (d, J = 8 Hz, CH(CH3)2, 2H), 1.17–1.19 (d, J = 8 Hz, CH(CH3)2, 30H), 1.54–1.56 (d, J = 8 Hz, CH(CH3)2, 2H), 2.12–2.14 (m, CH2–CH2, 6H), 2.26 (s, CH3, 6H), 2.26 (s, CH3, 6H), 2.31–2.33 (m, CH2–CH2, 2H), 2.33–2.52 (m, CH–N, 2H), 2.52–2.55 (d, J = 12 Hz, Ar-CH2, 2H), 3.21–3.24 (d, J = 12 Hz, N–CH2–N, 2H) 4.01–4.03 (d, J = 8 Hz, Ar-CH2, 2H), 4.22–4.27 (m, CH(CH3)2 (ter), 2H), 4.28–4.45 (m, CH(CH3)2 (ter), 2H), 4.48–4.71 (m, CH(CH3)2 (bri), 1H), 4.87–4.93 (m, CH(CH3)2 (bri), 1H), 6.50–6.54 (d, J = 16 Hz, Ar-H, 2H), 6.77 (s, Ar-H, 2H). 13C NMR (100 MHz, CDCl3): δ = 16.67 (CH3), 20.60 (CH3), 24.56 (CH2–CH2), 25.10 (CH2–CH2), 25.38 (CH2–CH2), 26.66 (CH2–CH2), 27.15 (CH(CH3)2), 27.48 (CH(CH3)2), 27.53 (CH(CH3)2), 28.53 (CH(CH3)2), 54.41 (CH–N), 59.56 (N–CH2–N) 63.93 (Ar-CH2), 70.46 (CH(CH3)2 (ter)), 71.17 (CH(CH3)2 (ter)), 72.07 (CH(CH3)2 (bri)), 72.44 (CH(CH3)2 (bri)), 122.87 (Ar-C), 123.37 (Ar-C), 125.31 (Ar-C), 128.02 (Ar-C), 131.36 (Ar-C), 158.11 (Ar-O). ESI m/z calculated for [M + Na]+. C43H74N2O8Zr2Na: 952.494 found 952.558. Anal. calcd for C43H74N2O8Zr2: C 54.22, H 7.83, N 2.94. Found: C 55.13, H 7.97, N 3.02.
Compound 2. Yield 0.07 g, (84%). Mp: 149 °C. 1H NMR (400 MHz, CDCl3): δ = 1.17–1.19 (d, J = 8 Hz, CH(CH3)2, 7H), 1.22–1.24 (d, J = 8 Hz, CH(CH3)2, 9H), 1.26–1.28 (d, J = 8 Hz, CH(CH3)2, 7H), 1.33–1.35 (d, J = 8 Hz, CH(CH3)2, 9H), 1.40 (s, C(CH3)3, 18H), 1.54–1.56 (d, J = 8 Hz, CH(CH3)2, 4H), 1.92–1.99 (m, CH2–CH2, 4H), 2.00–2.02 (m, CH2–CH2, 2H), 2.03 (s, CH3, 6H), 2.14–2.18 (m, CH2–CH2, 2H), 2.20–2.21 (m, CH–N, 2H), 3.22–3.25 (d, J = 12 Hz, Ar-CH2, 2H), 3.46–3.49 (d, J = 12 Hz, N–CH2–N, 2H), 4.09–4.12 (d, J = 12 Hz, Ar-CH2, 2H), 4.26–4.30 (m, CH(CH3)2 (ter), 2H), 4.30–4.43 (m, CH(CH3)2 (ter), 2H), 4.44–4.47 (m, CH(CH3)2 (bri), 1H), 4.49–4.69 (m, CH(CH3)2 (bri), 1H), 6.57–6.60 (d, J = 12 Hz, Ar-H, 2H), 6.89–6.92 (d, J = 12 Hz, Ar-H, 2H). 13C NMR (100 MHz, CDCl3): δ = 16.77 (CH3), 20.87 (CH2–CH2), 25.35 (CH2–CH2), 25.96 (CH2–CH2), 27.07 (CH2–CH2), 27.53 (CH(CH3)2), 29.50 (CH(CH3)2), 29.64 (CH(CH3)2), 30.79 (CH(CH3)2), 30.94 (C(CH3)3), 36.31 (C(CH3)3), 55.63 (CH–N), 58.49 (N–CH2–N), 64.93 (Ar-CH2), 68.19 (CH(CH3)2 (ter)), 71.06 (CH(CH3)2 (ter)), 71.26 (CH(CH3)2 (bri)), 72.48 (CH(CH3)2 (bri)), 125.46 (Ar-C), 126.93 (Ar-C), 128.39 (Ar-C), 128.60 (Ar-C), 129.20 (Ar-C), 160.00 (Ar-O). ESI m/z calculated for [M]+. C49H86N2O8Zr2: 1013.664 found 1013.691. Anal. calcd for C49H86N2O8Zr2: C 58.06, H 8.55, N 2.76. Found: C 57.93, H 8.94, N 2.61.
Compound 3. Yield 0.07 g, (86%). Mp: 153 °C. 1H NMR (400 MHz, CDCl3): δ = 0.88–0.90 (d, J = 8 Hz, CH(CH3)2, 4H), 1.19 (s, C(CH3)3, 18H), 1.22–1.24 (d, J = 8 Hz, CH(CH3)2, 26H), 1.32 (s, C(CH3)3, 18H), 1.48–1.50 (d, J = 8 Hz, CH(CH3)2, 6H), 1.56–1.93 (m, CH2–CH2, 2H), 2.09–2.12 (m, CH2–CH2, 2H), 2.27–2.28 (m, CH–N, 2H), 2.51–2.52 (m, CH2–CH2, 4H), 2.55–2.58 (d, J = 12 Hz, Ar-CH2, 2H), 3.23–3.26 (d, J = 12 Hz, Ar-CH2, 2H), 4.14–4.16 (d, J = 8 Hz, Ar-CH2, 2H), 4.27–4.32 (m, CH(CH3)2 (ter), 2H), 4.34–4.44 (m, CH(CH3)2 (ter), 2H), 4.45–4.51 (m, CH(CH3)2 (bri), 1H), 4.52–4.92 (m, CH(CH3)2 (bri), 1H), 6.71–6.75 (d, J = 16 Hz, Ar-H, 2H), 7.10–7.12 (d, J = 8 Hz, Ar-H, 2H). 13C NMR (100 MHz, CDCl3): δ = 22.84 (CH2–CH2), 24.66 (CH2–CH2), 24.97 (CH2–CH2), 25.42 (CH2–CH2), 27.39 (CH(CH3)2), 27.55 (CH(CH3)2), 28.34 (CH(CH3)2), 29.73 (CH(CH3)2), 30.85 (C(CH3)3), 31.76 (C(CH3)3), 33.96 (C(CH3)3), 35.21 (C(CH3)3), 54.63 (CH–N), 59.49 (N–CH2–N), 63.93 (Ar-CH2), 71.11 (CH(CH3)2 (ter)), 71.35 (CH(CH3)2 (ter)), 72.16 (CH(CH3)2 (bri)), 72.45 (CH(CH3)2 (bri)), 122.99 (Ar-C), 123.71 (Ar-C), 124.01 (Ar-C), 124.38 (Ar-C), 128.39 (Ar-C), 135.34 (Ar-C), 159.36 (Ar-O). ESI m/z calculated for [M + Na]+. C55H98N2O8Zr2Na: 1120.813 found 1120.089. Anal. calcd for C55H98N2O8Zr2: C 58.94, H 8.81, N 2.50. Found: C 59.09, H 8.71, N 2.45.
X-ray structure determination of compounds 1, 2 and 3
Suitable single crystals for X-ray diffraction studies were obtained from all the three compounds synthesized in this study. Single crystals were grown in a glove box at −24 °C from concentrated toluene solutions of the respective compounds over a period of 7 days. X-ray data was collected with a Bruker AXS (Kappa Apex 2) CCD diffractometer equipped with graphite monochromatic Mo (Kα) (λ = 0.7107 Å) radiation source. The data were collected with 100% completeness for θ up to 25°. ω and ϕ scans were employed to collect the data. The frame width for ω for was fixed to 0.5° for data collection. The frames were subjected to integration and data were reduced for Lorentz and polarization corrections using SAINT-NT. The multi-scan absorption correction was applied to the data set. All structures were solved using SIR-92 and refinement was done using SHELXL-97.78 Location of all the hydrogen atoms could be found in the difference Fourier map. The hydrogen atoms attached to carbon atoms were fixed at chemically meaningful positions and were allowed to ride with the parent atom during refinement. These data were deposited with CCDC with the following numbers: CCDC 980368, CCDC 1015322 and CCDC 980369. The crystal data is given in Table S1 (ESI†).
General procedure for the bulk polymerization of rac-LA and ε-CL
The polymerizations were done in 200:1 ratio between the respective monomers and compounds 1–3 under solvent free conditions. The polymerizations were performed by charging 236.6 μmol of 1–3 and 5.4 g (5 mL) (47.3 mmols) of ε-CL or 173.4 μmol of 1–3 and 5 g (34.7 mmols) of rac-LA under an argon atmosphere into a 100 mL glass-lined stainless steel autoclave reactor with mechanical stirring. The autoclave was heated to 80 °C for ε-CL and 140 °C in case of rac-LA. The reaction mixture was quenched by cooling the autoclave to ambient temperature in about 90 min and subsequently pouring the contents into cold heptane for ε-CL and cold methanol in case of rac-LA. The polymer was isolated by subsequent filtration and dried till a constant weight was attained. In presence of benzyl alcohol, 867.2 μmol of benzyl alcohol was added with 173.4 μmol of catalyst and 5 g (34.7 mmol) of rac-LA during the polymerization. For ε-CL polymerization 236.6 μmol of catalyst and 5.4 g (5 mL) (47.3 mmol) of ε-CL was added with 1183 μmol of benzyl alcohol during the ε-CL polymerization. The polymerization procedure was same as above. Cold methanol for rac-LA and cold heptane for ε-CL was used for the quenching of polymer. The formed polymer was filtered and dried in vacuum.
Kinetics of rac-LA polymerization
To determine the kinetics for the polymerization of rac-LA, we carried out the polymerizations reaction in small scale in a glass reactor at a temperature of 140 °C under argon atmosphere. At 600:1 ratio the polymerizations were performed by charging 11.56 μmol of 1–3 (11 mg of 1, 12 mg of 2 and 13 mg of 3) and 1 g (6.94 mmols) of rac-LA under an argon atmosphere. Next, the aliquots were taken out at regular time interval from the glass reactor under argon atmosphere and 1H NMR spectra were recorded to determine the % conversion of monomer into the corresponding polymer by comparing the methine proton of the unreacted monomer and polymer. Using this data we got linear plot by plotting ln([M]o/[M]t) vs. time. Apparent rate constant (kapp) were obtained from the slopes of the best fit lines. Next, we ran the GPC to get the molecular weight (Mn) and MWDs after quenching the aliquots removed after the same time intervals as above. From this we plotted Mn vs. % conversion vs. MWDs.
Kinetics of ε-CL polymerization
High Performance Liquid Chromatography (HPLC) was used for carrying out the kinetic investigation as it determines the concentration of various starting materials and product present as a function of time. To carry out the experiment we used reversed-phase C18 HPLC column and HPLC grade methanol as an eluting solvent. Before running the HPLC we sonicated the solvent for 30 min. The polymerizations were performed by charging 45.12 μmol of 1–3 (42 mg of 1, 46 mg of 2 and 50 mg of 3) and 1.03 g (1 mL) (9.02 mmols) of ε-CL under an argon atmosphere. We ran the HPLC after collecting the aliquots at regular time interval to determine the % conversion of monomer into the corresponding polymer. Using this data, we got linear plot by plotting ln([M]o/[M]t) vs. time. Apparent rate constant (kapp) were obtained from the slopes of the best fit lines. Next, we ran the GPC to determine the molecular weight (Mn) and MWDs after quenching the aliquots removed after the same time intervals as above. From this we plotted Mn vs. % conversion vs. MWDs.
General procedure for copolymerization reactions of epoxides (CHO and PO) and CO2
All the polymerization experiments were performed in a 100 mL stainless steel pressure vessel (Parr) equipped with an overhead stirrer. The autoclave was prepared by heating at 130 °C overnight to remove traces of moisture and cooled under vacuum to ambient temperature. The catalyst 1 (46 mg, 4.94 × 10−5 mol), 2 (50 mg, 4.94 × 10−5 mol) and 3 (54 mg, 4.94 × 10−5 mol), TBAB (16 mg, 1.0 molar ratio with respect to the catalyst), and CHO (5.0 mL, 4.85 g, 49.42 mmol, 1000 equiv with respect to catalyst) were transferred to the autoclave under an argon atmosphere. The autoclave was closed and charged with 35 bar CO2 pressure and heated to 50 °C. After 6 hours, the autoclave was allowed to cool to room temperature and vented in a fume hood. An aliquot for NMR was taken immediately after opening for the determination of yield and conversion. The resulting polycarbonate was dissolved in CH2Cl2 (5 mL), precipitated from MeOH (20 mL), collected and dried under vacuum at 100 °C to constant weight to determine the yield. In case of PO we used catalyst 1 (66 mg, 7.14 × 10−5 mol), 2 (72 mg, 7.14 × 10−5 mol) and 3 (78 mg, 7.14 × 10−5 mol), TBAB (23 mg, 1.0 molar ratio with respect to the catalyst), and PO (5.0 mL, 4.15 g, 71.45 mmol, 1000 equiv with respect to catalyst). Here, the autoclave was pressurized with CO2 to 35 bar at 40 °C for 6 hours. The same work-up procedure as above was followed.
General procedure for the coupling reaction of SO and CO2
For the coupling reaction of SO with CO2, the same procedure was followed as above. The catalyst 1 (41 mg, 4.38 × 10−5 mol), 2 (44 mg, 4.38 × 10−5 mol) and 3 (48 mg, 4.38 × 10−5 mol), TBAB (14 mg, 1.0 molar ratio with respect to the catalyst), and SO (5.0 mL, 5.26 g, 43.78 mmol, 1000 equiv with respect to catalyst) were transferred to the autoclave under an argon atmosphere. The autoclave was pressurized with CO2 to 35 bar at 80 °C for 8 hours. The same work-up procedure as above was followed.
General procedure for the homopolymerization of CHO, PO and SO
All the polymerization experiments were performed in a 100 mL stainless steel pressure vessel (Parr) equipped with an overhead stirrer. The autoclave was prepared by heating at 130 °C overnight to remove traces of moisture and the apparatus was cooled under vacuum to ambient temperature. The catalysts 1, 2 and 3 with CHO, PO or SO (1000 or 10000 equiv with respect to catalyst) were transferred to the autoclave under an argon atmosphere. The autoclave was shut and heated to 80 °C for CHO, 30 °C for PO and 120 °C for SO. After 6 hours (48 hours for 10000:1), the autoclave was allowed to cool to room temperature. An aliquot for NMR was taken immediately after opening for the determination of yield and conversion. The resulting polymer was dissolved in CH2Cl2 (5 mL), precipitated from acidic MeOH (20 mL), collected and dried under vacuum at 100 °C to constant weight to determine the yield.
General procedure for ethylene polymerization
The polymerizations were performed in a 100 mL stainless steel autoclave reactor with mechanical stirring under an argon atmosphere. The container was charged under an argon atmosphere with 50 mg of catalyst, 45 mL of hexane along with the required amount of MAO. Consequently, the autoclave was heated up to 50 °C and the ethylene gas was continuously bubbled through the proper channel. The gas feed was passed for 30 min at a pressure of 8 atm and subsequently the polymerization was quenched with acidic methanol. The polymer produced was collected by filtration and dried until a constant weight was achieved.
Characterization of polymers
Molecular weights (Mn) and the molecular weight distributions (MWDs) of the polymer samples produced by the ROP of various cyclic ester monomers and lactides were determined by using a GPC instrument with Waters 510 pump and Waters 410 Differential Refractometer as the detector. Three columns, WATERS STRYGEL-HR5, STRYGEL-HR4 and STRYGEL-HR3 each of dimensions (7.8 × 300 mm) were serially connected one after another. Measurements were done in THF at 27 °C for all the cases. Measurement of number average molecular weights (Mn), weight average molecular weights (Mw) and molecular weight distributions (Mw/Mn) (MWDs) of the polymers were performed relative to polystyrene standards. Molecular weights (Mn) were corrected according to Mark–Houwink corrections (Barakat et al., 1993). Molecular weights (Mn) and MWDs of the copolymer synthesized using epoxides and CO2 were determined in THF as the eluent at 27 °C, at a flow rate of 1 mL min−1 using polystyrene standards. Molecular weights (Mn and Mw) and the polydispersity indices (Mw/Mn) of polyethylene samples were obtained by a GPC instrument with Waters 510 pump and Waters 2414 differential refractometer as the detector. The column namely WATERS STRYGEL-HR4 of dimensions (4.6 × 300 mm) was fixed during the experiment. Measurements were performed in trichlorobenzene (TCB). Number average molecular weights (Mn) and molecular weight distributions (MWDs) of polymers were determined relative to polystyrene standards.
Differential scanning calorimetry
Glass transition (Tg) temperatures were measured using a DSC Q200 MDSC equipped with a refrigerated cooling system (RCS) with a working range of −90 to +20 °C. Samples (∼4 mg) were weighed into 40 μL aluminum pans and subjected to two heating cycles under nitrogen at a flow rate of 20 mL min−1. The first heating cycle consisted of heating from 0 to 100 °C at a rate of 10 °C min−1, held for 2 min at 100 °C and then cooled back to 0 at 10 °C min−1. The sample was held at this temperature for 2 min and subjected to a second heating cycle from 0 to 180 °C at a rate of 10 °C min−1.
Thermal gravimetric analysis
Thermal gravimetric analysis (TGA) was performed with a TA Instruments Q500. Under nitrogen atmosphere samples (5 mg) were loaded onto a platinum pan and subjected to a dynamic high-resolution scan, with an initial heating rate of 20 °C min−1. Each sample was heated from room temperature to 900 °C.
Computational details
All calculations have been performed employing Gaussian 09 quantum chemical program.61 Full geometry optimizations without any geometry constraint and frequency calculations on all the stationary points, were carried out using the Becke three parameter exchange functional in conjunction with Lee–Yang–Parr correlation functional (B3LYP), in combination with the LANL2DZ basis set.62 Molecular coordinates for the DFT calculations were extracted from single crystal XRD data. MPA (Mulliken Population Analysis) were performed using MPA 3.1 program as implemented in the Gaussian 09W package at the B3LYP/LANL2DZ level.62
Acknowledgements
The authors thank the Department of Science and Technology, New Delhi, for financial support.
Notes and references
-
(a) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC;
(b) H. Yasuda, Prog. Polym. Sci., 2000, 25, 573–626 CrossRef CAS;
(c) M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS;
(d) G. W. Coates, Dalton Trans., 2002, 467–475 RSC;
(e) M. H. Chisholm and Z. Zhou, J. Mater. Chem., 2004, 14, 3081–3092 RSC.
-
(a) M. J.-L. Tschan, E. Brulé, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC;
(b) J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS PubMed.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
-
(a) J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC;
(b) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed;
(c) K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed;
(d) C. Jerome and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef CAS PubMed;
(e) S. Penczek, M. Cypryk, A. Duda, P. Kubisa and S. Slomkowski, Prog. Polym. Sci., 2007, 32, 247–282 CrossRef CAS PubMed;
(f) R. Langer and J. P. Vacanti, Science, 1993, 260, 920–926 CAS.
-
(a) D. Li, Y. Peng, C. Geng, K. Liu and D. Kong, Dalton Trans., 2013, 42, 11295–11303 RSC;
(b) S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45, 4783–4790 CrossRef CAS PubMed.
- Y. Huang, W. Wang, C.-C. Lin, M. P. Blake, L. Clark, A. D. Schwarz and P. Mountford, Dalton Trans., 2013, 42, 9313–9324 RSC.
-
(a) W. Yi and H. Ma, Dalton Trans., 2014, 43, 5200–5210 RSC;
(b) Y. Wang, B. Liu, X. Wang, W. Zhao, D. Liu, X. Liu and D. Cui, Polym. Chem., 2014, 5, 4580–4588 RSC;
(c) Y. Gao, Z. Dai, J. Zhang, X. Ma, N. Tang and J. Wu, Inorg. Chem., 2014, 53, 716–726 CrossRef CAS PubMed;
(d) S. Song, H. Ma and Y. Yang, Dalton Trans., 2013, 42, 14200–14211 RSC;
(e) L. F. Sánchez-Barba, A. Garcés, J. Fernández-Baeza, A. Otero, C. Alonso-Moreno, A. Lara-Sánchez and A. M. Rodríguez, Organometallics, 2011, 30, 2775–2789 CrossRef.
- M. H. Chisholm, J. C. Huffman and K. Phomphrai, J. Chem. Soc., Dalton Trans., 2001, 222–224 RSC.
- C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832–4846 RSC.
- V. Poirier, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2009, 9820–9827 RSC.
-
(a) X.-F. Yu, C. Zhang and Z.-X. Wang, Organometallics, 2013, 32, 3262–3268 CrossRef CAS;
(b) C.-H. Wang, C.-Y. Li, B.-H. Huang, C.-C. Lin and B.-T. Ko, Dalton Trans., 2013, 42, 10875–10884 RSC;
(c) R. Petrus and P. Sobota, Dalton Trans., 2013, 42, 13838–13844 RSC;
(d) T. K. Sen, A. Mukherjee, A. Modak, S. K. Mandal and D. Koley, Dalton Trans., 2013, 42, 1893–1904 RSC.
- M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845–11854 CrossRef CAS.
-
(a) Z. Mou, B. Liu, M. Wang, H. Xie, P. Li, L. Li, S. Li and D. Cui, Chem. Commun., 2014, 50, 11411–11414 RSC;
(b) S. Abbina and G. Du, ACS Macro Lett., 2014, 3, 689–692 CrossRef CAS PubMed.
- M. H. Chisholm, C.-C. Lin, J. C. Gallucci and B.-T. Ko, Dalton Trans., 2003, 406–412 RSC.
- Z. Zhong, P. J. Dijkstra, C. Birg, M. Westerhausen and J. Feijen, Macromolecules, 2001, 34, 3863–3868 CrossRef CAS.
-
(a) M. Kuzdrowska, L. Annunziata, S. Marks, M. Schmid, C. G. Jaffredo, P. W. Roesky, S. M. Guillaume and L. Maron, Dalton Trans., 2013, 42, 9352–9360 RSC;
(b) B. Antelmann, M. H. Chisholm, S. S. Iyer, J. C. Huffman, D. Navarro-Llobet, M. Pagel, W. J. Simonsick and W. Zhong, Macromolecules, 2001, 34, 3159–3175 CrossRef CAS.
-
(a) C. T. Altaf, H. Wang, M. Keram, Y. Yang and H. Ma, Polyhedron, 2014, 81, 11–20 CrossRef CAS PubMed;
(b) S.-Y. Hsu, C.-H. Hu, C.-Y. Tu, C.-H. Lin, R.-Y. Chen, A. Datta and J.-H. Huang, Eur. J. Inorg. Chem., 2014, 1965–1973 CrossRef CAS;
(c) S. Tabthong, T. Nanok, P. Kongsaeree, S. Prabpai and P. Hormnirun, Dalton Trans., 2014, 43, 1348–1359 RSC;
(d) J. Liu and H. Ma, Dalton Trans., 2014, 43, 9098–9110 RSC;
(e) H.-L. Han, Y. Liu, J.-Y. Liu, K. Nomurac and Y.-S. Li, Dalton Trans., 2013, 42, 12346–12353 RSC;
(f) M. P. F. Pepels, M. Bouyahyi, A. Heise and R. Duchateau, Macromolecules, 2013, 46, 4324–4334 CrossRef CAS;
(g) K. Bakthavachalam and N. D. Reddy, Organometallics, 2013, 32, 3174–3184 CrossRef CAS;
(h) E. D. Cross, L. E. N. Allan, A. Decken and M. P. Shaver, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1137–1146 CrossRef CAS;
(i) A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114–2122 CrossRef CAS;
(j) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS PubMed.
-
(a) C.-X. Cai, A. Amgoune, C. W. Lehmann and J.-F. Carpentier, Chem. Commun., 2004, 330–331 RSC;
(b) T.-P.-A. Cao, A. Buchard, X. F. L. Goff, A. Auffrant and C. K. Williams, Inorg. Chem., 2012, 51, 2157–2169 CrossRef CAS PubMed;
(c) C.-Y. Tsai, H.-C. Du, J.-C. Chang, B.-H. Huang, B.-T. Ko and C.-C. Lin, RSC Adv., 2014, 4, 14527–14537 RSC.
-
(a) G. R. Giesbrecht, G. D. Whitener and J. Arnold, J. Chem. Soc., Dalton Trans., 2001, 923–927 RSC;
(b) H. E. Dyer, S. Huijser, A. D. Schwarz, C. Wang, R. Duchateau and P. Mountford, Dalton Trans., 2008, 32–35 RSC;
(c) F. Bonnet, A. R. Cowley and P. Mountford, Inorg. Chem., 2005, 44, 9046–9055 CrossRef CAS PubMed;
(d) H. Ma and J. Okuda, Macromolecules, 2005, 38, 2665–2673 CrossRef CAS.
-
(a) E. J. Lee, K. M. Lee, J. Jang, E. Kim, J. S. Chunga, Y. Do, S. C. Yoon and S. Y. Park, J. Mol. Catal. A: Chem., 2014, 385, 68–72 CrossRef CAS PubMed;
(b) L. Wang, V. Poirier, F. Ghiotto, M. Bochmann, R. D. Cannon, J.-F. Carpentier and Y. Sarazin, Macromolecules, 2014, 47, 2574–2584 CrossRef CAS.
-
(a) A.-L. Mogstad and R. M. Waymouth, Macromolecules, 1994, 27, 2313–2315 CrossRef CAS;
(b) C. Miola-Delaite, T. Hamaide and R. Spitz, Macromol. Chem. Phys., 1999, 200, 1771–1778 CrossRef CAS;
(c) D. Takeuchi, T. Nakamura and T. Aida, Macromolecules, 2000, 33, 725–729 CrossRef CAS;
(d) Y. Kim and J. G. Verkade, Macromol. Rapid Commun., 2002, 23, 917–921 CrossRef CAS;
(e) Y. Kim and J. G. Verkade, Organometallics, 2002, 21, 2395–2399 CrossRef CAS;
(f) Y. Takashima, Y. Nakayama, T. Hirao, H. Yasuda and A. Harada, J. Organomet. Chem., 2004, 689, 612–619 CrossRef CAS PubMed;
(g) P. Dobrzynski, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1886–1900 CrossRef CAS;
(h) P. Dobrzynski, S. Li, J. Kasperczyk, M. Bero, F. Gasc and M. Vert, Biomacromolecules, 2005, 6, 483–488 CrossRef CAS PubMed;
(i) Y. Kim and J. G. Verkade, Macromol. Symp., 2005, 224, 105–118 CrossRef CAS;
(j) C. K. A. Gregson, I. J. Blackmore, V. C. Gibson, N. J. Long, E. L. Marshall and A. J. P. White, Dalton Trans., 2006, 3134–3140 RSC;
(k) D. Patel, S. T. Liddle, S. A. Mungur, M. Rodden, A. Blake and P. L. Arnold, Chem. Commun., 2006, 1124–1126 RSC;
(l) J. Cheng, J. Sun, K. Wu and X. Yan, Huaxue Gongye Yu Gongcheng Jishu, 2006, 27, 5–7 CAS;
(m) A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn, M. F. Mahon, A. F. Johnson, P. Khunkamchoo, S. L. Roberts and S. S. F. Wong, Macromolecules, 2006, 39, 7250–7257 CrossRef CAS;
(n) A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Dalton Trans., 2006, 887–889 RSC;
(o) K. C. Hsieh, W. Y. Lee, L. F. Hsueh, H. M. Lee and J. H. Huang, Eur. J. Inorg. Chem., 2006, 2306–2312 CrossRef CAS;
(p) C. J. Chuck, M. G. Davidson, M. D. Jones, G. Kociok-Köhn, M. D. Lunn and S. Wu, Inorg. Chem., 2006, 45, 6595–6597 CrossRef CAS PubMed;
(q) Y. Sarazin, R. H. Howard, D. L. Hughes, S. M. Humphrey and M. Bochmann, Dalton Trans., 2006, 340–350 RSC;
(r) F. Gornshtein, M. Kapon, M. Botoshansky and M. Eisen, Organometallics, 2007, 26, 497–507 CrossRef CAS;
(s) J. Lee, Y. Kim and Y. Do, Inorg. Chem., 2007, 46, 7701–7703 CrossRef CAS PubMed.
-
(a) A. J. Chmura, D. M. Cousins, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Dalton Trans., 2008, 1437–1443 RSC;
(b) A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293–1295 RSC;
(c) Y. Ning, Y. Zhang, A. R. Delgado and E. Y.-X. Chen, Organometallics, 2008, 27, 5632–5640 CrossRef CAS;
(d) A. Grafov, S. Vuorinen, T. Repo, M. Kemell, M. Nieger and M. Leskelä, Eur. Polym. J., 2008, 44, 3797–3805 CrossRef CAS PubMed;
(e) A. L. Zelikoff, J. Kopilov, I. Goldberg, G. W. Coates and M. Kol, Chem. Commun., 2009, 6804–6806 RSC;
(f) A. D. Schwarz, A. L. Thompson and P. Mountford, Inorg. Chem., 2009, 48, 10442–10454 CrossRef CAS PubMed;
(g) E. Kim, E. W. Shin, I.-K. Yoo and J. S. Chung, J. Mol. Catal. A: Chem., 2009, 298, 36–39 CrossRef CAS PubMed;
(h) E. L. Whitelaw, M. D. Jones, M. F. Mahon and G. Kociok-Köhn, Dalton Trans., 2009, 9020–9025 RSC;
(i) M. D. Jones, M. G. Davidson and G. Kociok-Köhn, Polyhedron, 2010, 29, 697–700 CrossRef CAS PubMed;
(j) F. Zhang, H. Song and G. Zi, J. Organomet. Chem., 2010, 695, 1993–1999 CrossRef CAS PubMed;
(k) E. Sergeeva, J. Kopilov, I. Goldberg and M. Kol, Inorg. Chem., 2010, 49, 3977–3979 CrossRef CAS PubMed;
(l) A. D. Schwarz, K. R. Herbert, C. Paniagua and P. Mountford, Organometallics, 2010, 29, 4171–4188 CrossRef CAS;
(m) M. Hu, M. Wang, H. Zhu, L. Zhang, H. Zhang and L. Sun, Dalton Trans., 2010, 39, 4440–4446 RSC;
(n) A. Stopper, I. Goldberg and M. Kol, Inorg. Chem. Commun., 2011, 14, 715–718 CrossRef CAS PubMed;
(o) S. L. Hancock, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 40, 2033–2037 RSC;
(p) C. Romain, B. Heinrich, S. B. Laponnaz and S. Dagorne, Chem. Commun., 2012, 48, 2213–2215 RSC;
(q) A. Stopper, J. Okuda and M. Kol, Macromolecules, 2012, 45, 698–704 CrossRef CAS.
-
(a) X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS PubMed;
(b) F. D. Monica, E. Luciano, G. Roviello, A. Grassi, S. Milione and C. Capacchione, Macromolecules, 2014, 47, 2830–2841 CrossRef;
(c) M. J. Go, J. M. Lee, K. M. Lee, C. H. Oh, K. H. Park, S. H. Kim, M. Kim, H.-R. Park, M. H. Park, Y. Kim and J. Lee, Polyhedron, 2014, 67, 286–294 CrossRef CAS PubMed;
(d) H.-W. Ou, H.-Y. Chen, H.-C. Tseng, M.-W. Hsiao, Y.-L. Chang, N.-Y. Jheng, Y.-C. Lai, T.-Y. Shih, Y.-T. Lin and H.-Y. Chen, J. Mol. Catal. A: Chem., 2014, 394, 97–104 CrossRef CAS PubMed;
(e) H.-J. Chuang, B.-H. Wu, C.-Y. Li and B.-T. Ko, Eur. J. Inorg. Chem., 2014, 1239–1248 CrossRef CAS;
(f) A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC;
(g) F. Marchetti, G. Pampaloni, C. Pinzino, F. Renili, T. Repo and S. Vuorinenc, Dalton Trans., 2013, 42, 2792–2802 RSC;
(h) A. Sauer, J.-C. Buffet, T. P. Spaniol, H. Nagae, K. Mashima and J. Okuda, Inorg. Chem., 2012, 51, 5764–5770 CrossRef CAS PubMed;
(i) L.-C. Liang, S.-T. Lin, C.-C. Chien and M.-T. Chen, Dalton Trans., 2013, 42, 9286–9293 RSC;
(j) R. L. Webster, N. Noroozi, S. G. Hatzikiriakos, J. A. Thomsona and L. L. Schafer, Chem. Commun., 2013, 49, 57–59 RSC;
(k) C.-Y. Li, C.-J. Yu and B.-T. Ko, Organometallics, 2013, 32, 172–180 CrossRef CAS;
(l) I. El-Zoghbi, T. J. J. Whitehorne and F. Schaper, Dalton Trans., 2013, 42, 9376–9387 RSC;
(m) R. R. Gowda and E. Y.-X. Chen, Dalton Trans., 2013, 42, 9263–9273 RSC;
(n) S. Pappuru, E. R. Chokkapu, D. Chakraborty and V. Ramkumar, Dalton Trans., 2013, 42, 16412–16427 RSC;
(o) D. Chakraborty, D. Mandal, V. Ramkumar, V. Subramanian and J. V. Sundar, Polymer, 2015, 56, 157–170 CrossRef CAS PubMed.
-
(a) T. K. Saha, M. Mandal, M. Thunga, V. Ramkumar and D. Chakraborty, Dalton Trans., 2013, 42, 10304–10314 RSC;
(b) Y. Kim, P. N. Kapoor and J. G. Verkade, Inorg. Chem., 2002, 41, 4834–4838 CrossRef CAS PubMed.
- A. J. Chmura, C. J. Chuck, M. G. Davidson, M. D. Jones, M. D. Lunn, S. D. Bull and M. F. Mahon, Angew. Chem., Int. Ed., 2007, 46, 2280–2283 CrossRef CAS PubMed.
-
(a) D. C. Aluthge, B. O. Patrick and P. Mehrkhodavandi, Chem. Commun., 2013, 49, 4295–4297 RSC;
(b) A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem., Int. Ed., 2008, 47, 2290–2293 CrossRef CAS PubMed.
- B. J. O'Keefe, S. M. Monnier, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2001, 123, 339–340 CrossRef.
- B. J. O'Keefe, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2002, 124, 4384–4393 CrossRef PubMed.
- D. K. Gilding and A. M. Read, Polymer, 1979, 20, 1459–1464 CrossRef CAS.
-
(a) Y. Sarazin, M. Schormann and M. Bochmann, Organometallics, 2004, 23, 3296–3302 CrossRef CAS;
(b) Y. Wang, W. Zhao, X. Liu, D. Cui and E. Y.-X. Chen, Macromolecules, 2012, 45, 6957–6965 CrossRef CAS;
(c) H.-Y. Chen, H.-Y. Tang and C.-C. Lin, Polymer, 2007, 48, 2257–2262 CrossRef CAS PubMed;
(d) P. Radano, G. L. Baker and M. R. Smith III, J. Am. Chem. Soc., 2000, 122, 1552–1553 CrossRef;
(e) S. Ghosh, D. Chakraborty and B. Varghese, Eur. Polym. J., 2015, 62, 51–65 CrossRef CAS PubMed.
-
(a) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS;
(b) M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC;
(c) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC;
(d) M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- P. Dubois, N. Ropson, R. Jérôme and P. Teyssié, Macromolecules, 1996, 29, 1965–1975 CrossRef CAS.
- M. Myers, E. F. Connor, T. Glauser, A. Möck, G. Nyce and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 844–851 CrossRef CAS.
-
(a) X. Wang, K. Liao, D. Quan and Q. Wu, Macromolecules, 2005, 38, 4611–4617 CrossRef CAS;
(b) K. Shinno, M. Miyamoto, Y. Kimura, Y. Hirai and H. Yoshitome, Macromolecules, 1997, 30, 6438–6444 CrossRef CAS.
-
(a) V. Katiyar and H. Nanavati, Polym. Chem., 2010, 1, 1491–1500 RSC;
(b) D. Garlotta, J. Polym. Environ., 2001, 9(2), 63–84 CrossRef CAS.
- B. L. Small and M. Brookhart, Macromolecules, 1999, 32, 2120–2130 CrossRef CAS.
-
(a) T. K. Saha, M. Mandal, D. Chakraborty and V. Ramkumar, New J. Chem., 2013, 37, 949–960 RSC;
(b) T. K. Saha, V. Ramkumar and D. Chakraborty, Inorg. Chem., 2011, 50, 2720–2722 CrossRef CAS PubMed.
- H. Arakawa, et al., Chem. Rev., 2001, 101, 953–996 CrossRef CAS PubMed.
-
(a) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed;
(b) G. W. Coates and D. R. Moore, Angew. Chem., 2004, 116, 6784–6806 CrossRef;
(c) H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573 CrossRef CAS;
(d) K. Nozaki, Pure Appl. Chem., 2004, 76, 541–546 CrossRef CAS;
(e) A. Rokicki and W. Kuran, J. Macromol.
Sci., Rev. Macromol. Chem. Phys., 1981, C21, 135–186 CAS.
-
(a) M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC;
(b) P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS;
(c) W. Leitner, Coord. Chem. Rev., 1996, 153, 257–284 CrossRef CAS.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
-
(a) T. Aida and S. Inoue, J. Am. Chem. Soc., 1983, 105, 1304–1309 CrossRef CAS;
(b) T. Aida, M. Ishikawa and S. Inoue, Macromolecules, 1986, 19, 8–13 CrossRef CAS;
(c) M. H. Chisholm and Z. P. Zhou, J. Am. Chem. Soc., 2004, 126, 11030–11039 CrossRef CAS PubMed;
(d) D. J. Darensbourg and D. R. Billodeaux, Inorg. Chem., 2005, 44, 1433–1442 CrossRef CAS PubMed;
(e) K. Nishioka, H. Goto and H. Sugimoto, Macromolecules, 2012, 45, 8172–8192 CrossRef CAS.
- G. A. Luinstra, Polym. Rev., 2008, 48, 192–219 CrossRef CAS.
- M. Chasin and R. Langer, Biodegradable Polymers as Drug Delivery Systems, Marcel Dekker Inc, New York, 1990 Search PubMed.
- W. Kuran, Prog. Polym. Sci., 1998, 23, 919–992 CrossRef CAS.
-
(a) D. J. Darensbourg and W.-C. Chung, Macromolecules, 2014, 47, 4943–4948 CrossRef CAS;
(b) D. J. Darensbourg and A. D. Yeung, Polym. Chem., 2014, 5, 3949–3962 RSC;
(c) C. Robert, T. Ohkawara and K. Nozaki, Chem.–Eur. J., 2014, 20, 4789–4795 CrossRef CAS PubMed;
(d) M. I. Childers, J. M. Longo, N. J. van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed;
(e) D. J. Darensbourg, R. M. Mackiewicz, A. M. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836–844 CrossRef CAS PubMed.
-
(a) T. Aida and S. Inoue, Acc. Chem. Res., 1996, 29, 39–48 CrossRef CAS;
(b) F. Kojima, T. Aida and S. Inoue, J. Am. Chem. Soc., 1986, 108, 391–395 CrossRef CAS PubMed;
(c) H. Sugimoto, H. Ohtsuka and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4172–4186 CrossRef CAS.
-
(a) T. Sarbu and E. J. Beckman, Macromolecules, 1999, 32, 6904–6912 CrossRef CAS;
(b) D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2599–2602 CrossRef CAS;
(c) M. Kroger, C. Folli, O. Walter and M. Doring, Adv. Synth. Catal., 2005, 347, 1325–1328 CrossRef;
(d) Y. L. Xiao, Z. Wang and K. L. Ding, Chem.–Eur. J., 2005, 11, 3668–3678 CrossRef CAS PubMed;
(e) W. C. Ellis, Y. Jung, M. Mulzer, R. D. Girolamo, E. B. Lobkovsky and G. W. Coates, Chem. Sci., 2014, 5, 4004–4011 RSC.
-
(a) R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498–11499 CrossRef CAS;
(b) S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 2000, 33, 303–308 CrossRef CAS.
-
(a) D. J. Darensbourg, J. C. Yarbrough, C. Ortiz and C. C. Fang, J. Am. Chem. Soc., 2003, 125, 7586–7591 CrossRef CAS PubMed;
(b) R. Eberhardt, M. Allmendinger and B. Rieger, Macromol. Rapid Commun., 2003, 24, 194–196 CrossRef CAS;
(c) D. J. Darensbourg and R. M. Mackiewicz, J. Am. Chem. Soc., 2005, 127, 14026–14038 CrossRef CAS PubMed;
(d) D. J. Darensbourg, R. M. Mackiewicz and D. R. Billodeaux, Organometallics, 2005, 24, 144–148 CrossRef CAS.
-
(a) B. Li, R. Zhang and X. B. Lu, Macromolecules, 2007, 40, 2303–2307 CrossRef CAS;
(b) X. Q. Xu, C. M. Wang, H. R. Li, Y. Wang, W. L. Sun and Z. Q. Shen, Polymer, 2007, 48, 3921–3924 CrossRef CAS PubMed;
(c) B. Li, G. P. Wu, W. M. Ren, Y. M. Wang, D. Y. Rao and X. B. Lu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6102–6113 CrossRef CAS;
(d) D. J. Darensbourg and S. B. Fitch, Inorg. Chem., 2009, 48, 8668–8677 CrossRef CAS PubMed;
(e) D. J. Darensbourg, M. Ulusoy, O. Karroonnirum, R. R. Poland, J. H. Reibenspies and B. Cetinkaya, Macromolecules, 2009, 42, 6992–6998 CrossRef CAS;
(f) K. Nakano, M. Nakamura and K. Nozaki, Macromolecules, 2009, 42, 6972–6980 CrossRef CAS;
(g) D. J. Darensbourg, R. R. Poland and A. L. Strickland, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 127–133 CrossRef CAS;
(h) L. P. Guo, C. M. Wang, W. J. Zhao, H. R. Li, W. L. Sun and Z. Q. Shen, Dalton Trans., 2009, 5406–5410 RSC.
-
(a) C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS PubMed;
(b) C. T. Cohen and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5182–5191 CrossRef CAS;
(c) M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef CAS;
(d) B. Liu, Y. Gao, X. Zhao, W. Yan and X. Wang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 359–365 CrossRef CAS;
(e) B. Liu, X. Zhao, H. Guo, Y. Gao, M. Yang and X. Wang, Polymer, 2009, 50, 5071–5075 CrossRef CAS PubMed;
(f) X. B. Lu and Y. Wang, Angew. Chem., Int. Ed., 2004, 43, 3574–3577 CrossRef CAS PubMed;
(g) K. Nakano, S. Hashimoto, M. Nakamura, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2011, 50, 4868–4871 CrossRef CAS PubMed;
(h) K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369–373 RSC;
(i) K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2006, 45, 7274–7277 CrossRef CAS PubMed;
(j) Y. S. Niu, H. C. Li, X. S. Chen, W. X. Zhang, X. Zhuang and X. B. Jing, Macromol. Chem. Phys., 2009, 210, 1224–1229 CrossRef CAS;
(k) Y. S. Niu, W. X. Zhang, X. Pang, X. S. Chen, X. L. Zhuang and X. B. Jing, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5050–5056 CrossRef CAS;
(l) E. K. Noh, S. J. Na, S. Sujith, S.-W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082–8083 CrossRef CAS PubMed;
(m) R. L. Paddock and S. T. Nguyen, Macromolecules, 2005, 38, 6251–6253 CrossRef CAS;
(n) Z. Q. Qin, C. M. Thomas, S. Lee and G. W. Coates, J. Am. Chem. Soc., 2003, 42, 5484–5487 CAS;
(o) W.-M. Ren, Z.-W. Liu, Y.-Q. Wen, R. Zhang and X.-B. Lu, J. Am. Chem. Soc., 2009, 131, 11509–11518 CrossRef CAS PubMed;
(p) W. M. Ren, X. Zhang, Y. Liu, J. F. Li, H. Wang and X. B. Lu, Macromolecules, 2010, 43, 1396–1402 CrossRef CAS;
(q) J. E. Seong, S. J. Na, A. Cyriac, B.-W. Kim and B. Y. Lee, Macromolecules, 2010, 43, 903–908 CrossRef CAS;
(r) G.-P. Wu, D. J. Darensbourg and X.-B. Lu, J. Am. Chem. Soc., 2012, 134, 17739–17745 CrossRef CAS PubMed.
-
(a) A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC;
(b) M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS PubMed.
-
(a) K. Nakano, K. Kobayashi and K. Nozaki, J. Am. Chem. Soc., 2011, 133, 10720–10723 CrossRef CAS PubMed;
(b) C. C. Quadri and E. L. Roux, Dalton Trans., 2014, 43, 4242–4246 RSC;
(c) H.-J. Chuang and B.-T. Ko, Dalton Trans., 2015, 44, 598–607 RSC;
(d) C.-K. Su, H.-J. Chuang, C.-Y. Li, C.-Y. Yu, B.-T. Ko, J.-D. Chen and M.-J. Chen, Organometallics, 2014, 33, 7091–7100 CrossRef CAS.
-
(a) D. J. Darensbourg, W.-C. Chung and S. J. Wilson, ACS Catal., 2013, 3, 3050–3057 CrossRef CAS;
(b) D. Tian, B. Liu, Q. Gan, H. Li and D. J. Darensbourg, ACS Catal., 2012, 2, 2029–2035 CrossRef CAS;
(c) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC;
(d) D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765–10780 CrossRef CAS PubMed.
- X. B. Lu, L. Shi, Y. M. Wang, R. Zhang, Y. J. Zhang, X. J. Peng, Z. C. Zhang and B. Li, J. Am. Chem. Soc., 2006, 128, 1664–1674 CrossRef CAS PubMed.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- T. Bok, H. Yun and B. Y. Lee, Inorg. Chem., 2006, 45, 4228–4237 CrossRef CAS PubMed.
-
(a) X. Xu, Y. Yan, Y. Zhang and Q. Shen, Inorg. Chem., 2007, 46, 3743–3751 CrossRef CAS PubMed.
-
(a) R. R. Gowda and D. Chakraborty, J. Mol. Catal. A: Chem., 2009, 301, 84–92 CrossRef CAS PubMed;
(b) A. Cohen, J. Kopilov, I. Goldberg and M. Kol, Organometallics, 2009, 28, 1391–1405 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark and J. J. Heyd, GAUSSIAN 09, Revision C.01, E. Brothers CT, 201 Search PubMed.
- I. A. Tonks, L. M. Henling, M. W. Day and J. E. Bercaw, Inorg. Chem., 2009, 48, 5096–5105 CrossRef CAS PubMed.
- D. Cheshmedzhieva, I. Angelova, S. Ilieva, G. S. Georgiev and B. Galabov, Comput. Theor. Chem., 2012, 995, 8–16 CrossRef CAS PubMed.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
-
(a) S. S. Natanael, R. B. Lima, A. L. P. Silva, A. A. Tanaka, A. B. F. Silva and J. J. G. Varela Jr, Comput. Theor. Chem., 2015, 1054, 93–99 CrossRef PubMed;
(b) T. M. Pappenfus, B. J. Hermanson, T. J. Helland, G. G. W. Lee, S. M. Drew, K. R. Mann, K. A. McGee and S. C. Rasmussen, Org. Lett., 2008, 10, 1553–1556 CrossRef CAS PubMed.
- S. I. Vagin, R. Reichardt, S. Klaus and B. Rieger, J. Am. Chem. Soc., 2010, 132, 14367–14369 CrossRef CAS PubMed.
- G. A. Luinstra, G. R. Haas, F. Molnar, V. Bernhart, R. Eberhardt and B. Rieger, Chem.–Eur. J., 2005, 11, 6298–6314 CrossRef CAS PubMed.
- K. Nakano, K. Nozaki and T. Hiyama, Macromolecules, 2001, 34, 6325–6332 CrossRef CAS.
- M. Kröger, C. Folli, O. Walter and M. Döring, Adv. Synth. Catal., 2006, 348, 1908–1918 CrossRef.
- R. K. Dean, L. N. Dawe and C. M. Kozak, Inorg. Chem., 2012, 51, 9095–9103 CrossRef CAS PubMed.
- S. D. Allen, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 14284–14285 CrossRef CAS PubMed.
-
(a) D. J. Darensbourg and A. D. Yeung, Macromolecules, 2013, 46, 83–95 CrossRef CAS;
(b) R. K. Dean, K. Devaine-Pressing, L. N. Dawe and C. M. Kozak, Dalton Trans., 2013, 42, 9233–9244 RSC;
(c) D. J. Darensbourg and S.-H. Wei, Macromolecules, 2012, 45, 5916–5922 CrossRef CAS;
(d) G.-P. Wu, S.-H. Wei, X.-B. Lu, W.-M. Ren and D. J. Darensbourg, Macromolecules, 2010, 43, 9202–9204 CrossRef CAS.
- B. Lin and D. L. Whalen, J. Org. Chem., 1994, 59, 1638–1641 CrossRef CAS.
- R. M. Thomas, P. C. B. Widger, S. M. Ahmed, R. C. Jeske, W. Hirahata, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 16520–16525 CrossRef CAS PubMed.
- W. Hirahata, R. M. Thomas, E. B. Lobkovsky and G.
W. Coates, J. Am. Chem. Soc., 2008, 130, 17658–17659 CrossRef CAS PubMed.
-
(a) J. Tian, P. D. Hustad and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 5134–5135 CrossRef CAS;
(b) J. Huang, B. Lian, L. Yong and Y. Qian, Inorg. Chem. Commun., 2001, 4, 392–394 CrossRef CAS;
(c) Y. Takii, P. M. Gurubasavaraj, S. Katao and K. Nomura, Organometallics, 2012, 31, 8237–8248 CrossRef CAS.
-
(a) K. Kawai and T. Fujita, Top. Organomet. Chem., 2009, 26, 3–46 CrossRef CAS;
(b) H. Makio, N. Kashiwa and T. Fujita, Adv. Synth. Catal., 2002, 344, 477–493 CrossRef CAS.
- G. M. Sheldrick, SHELXL97. Program for crystal structure Refinement, Göttingen, Germany Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full spectroscopic, crystallographic, polymerization data, kinetics for polymerization, and mechanism for the concerned polymerization data are given in ESI. CCDC 980368, 1015322 and 980369. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra17201a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.