
Rational design, synthesis, and 2D-QSAR study of anti-oncological alkaloids against hepatoma and cervical carcinoma†
Abstract
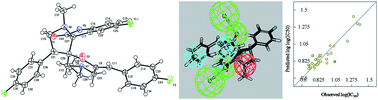
Antitumor active dispiro[3H-indole-3,2′-pyrrolidine-3′,3′′-piperidine]-2(1H),4′′-diones 11–19 were regioselectively synthesized via azomethine ylide cycloaddition reactions with 3E,5E-1-alkyl-3,5-bis(arylmethylidene)-4-piperidones 3–7. Compounds 13, 14, and 16 reveal higher potency against the HeLa (cervical) tumor cell line than the standard reference cisplatin, while 11, and 12 seem more potent against the HepG2 (liver) carcinoma cell line relative to the standard reference doxorubicin hydrochloride as determined by in vitro Sulfo-Rhodamine-B bio-assay. 3D-Pharmacophores of the HeLa comprise five chemical features viz., two hydrogen bond acceptors, two hydrophobic centers and one positive ionizable center and HepG2 contains three chemical features viz., a hydrogen bond acceptor, a hydrophobic center and a positive ionizable center. These features of the tumor cell lines explain the variation of bioactivity relative to chemical structure. Statistically significant QSAR models describing the spiro-alkaloid bio-properties were obtained employing CODESSA-Pro software validating the observed pharmacological observations and identifying the most important parameters governing activity.
 Please wait while we load your content...
Please wait while we load your content...

