
The role of hydroxyl channel in defining selected physicochemical peculiarities exhibited by hydroxyapatite
Abstract
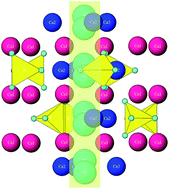
Mysteries surrounding the most important mineral for vertebrate biology, hydroxyapatite, are many. Perhaps the Greek root of its name, απαταo, meaning ‘to deceive’ and given to its mineral form by the early gem collectors who confused it with more precious stones, is still applicable today, though in a different connotation, descriptive of a number of physicochemical peculiarities exhibited by it. Comparable to water as the epitome of peculiarities in the realm of liquids, hydroxyapatite can serve as a paradigm of peculiarities in the world of solids. Ten of the peculiar properties of hydroxyapatite are sketched in this study, ranging from (i) the crystal lattice flexibility to (ii) notorious surface layer instability, (iii) finite piezoelectricity, pyroelectricity and conductivity to protons, (iv) accelerated growth and improved osteoconductivity in electromagnetic fields, (v) a high nucleation rate at low supersaturation and low crystal growth rate at high supersaturation, (vi) higher bioactivity and resorbability of biological apatite compared to those of the synthetic ones, and beyond. An attempt has been made to explain this array of curious characteristics by referring to a particular element of the crystal structure of hydroxyapatite: the hydroxyl ion channel extending in the direction of the c-axis, through a crystallographic column created by the overlapping calcium ion triangles.
 Please wait while we load your content...
Please wait while we load your content...

