
Performance studies of PEI/SPEI blend ultra-filtration membranes via surface modification using cSMM additives
Abstract
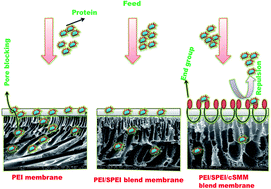
Sulfonated poly(ether imide) (SPEI) and charged surface modifying macromolecules (cSMM) were synthesized, characterized and blended into the casting solution of poly (ether imide) (PEI) with different compositions to develop surface modified ultra-filtration (UF) membranes by means of improved hydrophilicity. The membranes were prepared by a phase inversion technique and subjected to characterization experiments such as water permeation tests, equilibrium water content (WC), membrane hydraulic resistance (Rm) and protein rejection. Among others, a blend membrane containing 20 wt% SPEI and 5 wt% cSMM in PEI (called M12) exhibited the highest water permeation (440.6 L m−2 h−1 at 345 kPa), highest WC (86.3%) and lowest Rm (0.7 kPa/L m−2 h−1). The M12 membrane also exhibited the highest fluxes of 364.1 L m−2 h−1 and 230.3 L m−2 h−1 (at 345 kPa) with rejections of 31.3% and 62.1%, respectively, when the feed was aqueous trypsin and bovine serum albumin solution (1000 ppm). The difference in contact angle between the top and bottom surface confirmed surface migration of cSMM to the membrane top surface. SEM, AFM and tensile strength measurement revealed that the surface became more porous and rougher, and the mechanical strength was lowered by the blending of SPEI and addition of cSMM. That the M12 membrane achieved a lower internal fouling of 6% and 3.8% and higher flux recovery ratio (FRR) of 94% and 96.2% after UF of trypsin and bovine serum albumin solution explained its better antifouling properties as compared to a pristine PEI membrane.
 Please wait while we load your content...
Please wait while we load your content...

