
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolysaccharide-assisted rapid exfoliation of graphite platelets into high quality water-dispersible graphene sheets†
Ilke Uysal Unalanab,
Chaoying Wanb,
Silvia Trabattonic,
Luciano Piergiovannia and
Stefano Farris*a
aDeFENS, Department of Food, Environmental and Nutritional Sciences—Packaging Division, University of Milan, Via Celoria 2, 20133 Milan, Italy. E-mail: stefano.farris@unimi.it; Fax: +39 0250316672; Tel: +39 0250316654
bInternational Institute for Nanocomposites Manufacturing, WMG, University of Warwick, Coventry CV4 7AL, UK
cDepartment of Materials Science, University of Milano Bicocca, Via Cozzi 55, 20125 Milan, Italy
First published on 9th March 2015
Abstract
Ultrasound exfoliation of graphite with the assistance of three polysaccharides (nonionic pullulan, cationic chitosan, and anionic alginate) was investigated in this work. The effects of polymer type, initial concentration of graphite, and ultrasonication period on the graphene yield and quality were compared. Under a sonotrode-type ultrasonication treatment for 30 min, graphene aqueous dispersions with concentrations of up to 2.3 mg ml−1 in pullulan solutions and 5.5 mg ml−1 in chitosan solutions were achieved. The obtained graphene nanosheets were characterized as low-defect mono-layer, bi-layer, and few-layer (<5), and formed stable dispersions in water for up to 6 months. The adsorption of pullulan and chitosan biopolymers on the graphene surface as determined by TGA technique was approximately 2.5 wt% and 8.5 wt%, respectively, which accounts for the dispersibility and stability of the graphene sheets in water. Findings arising from this work suggest that pullulan and chitosan are more effective in exfoliating graphite into graphene than alginate due to the different surface free energy and thermodynamic affinity. The polysaccharide-assisted aqueous-exfoliation approach enables the production of water-dispersible graphene with high quality and large quantity, thus providing an industrially scalable route for new potential applications of graphene-based nanocomposites, e.g. in the food packaging industry.
Introduction
Due to its fascinating properties, graphene is one of today's most exciting materials, providing an unexpected performance that can be profitably used in many different fields.1,2 One of the most active fields of investigation concerns the development of polymer–graphene composites for a wide range of applications. While lab-scale experiments have successfully demonstrated the enhanced performance of graphene-based composites, large-scale utilization is often hindered due to the high production costs mainly associated with the highly expensive and low-yielding methods and procedures to obtain graphene monolayers from graphite.3 For this reason, the development of cheap, high-throughput, user-friendly, and possibly biocompatible new approaches is highly desirable to implement market applications. Moreover, decreasing production costs would make graphene readily usable also in low added value markets, e.g. food packaging, where graphene has recently been suggested as a potential filler due to its expected enhancement of mechanical,4–6 thermal,4,6–8 and barrier properties against O2,5–9 CO2,6 and water vapor4 of the final packaging materials (e.g., films and coatings).Production of graphene has been achieved by different methods, most falling within the top-down approach, such as: micromechanical cleavage of graphite, also known as the “Scotch tape method”;10 reduction of graphene oxide;11–13 thermal techniques;14–16 UV-assisted photocatalytic techniques;17,18 exfoliation of graphite by high shear mixing, such as ball milling19 and roll milling;20 and sonication.21 Although the low costs and high ease of processing and scalability, the main disadvantages linked to the top-down strategies are related to the poor quality output22,23 and the excessive use of harsh and aggressive reagents (e.g. H2SO4/KMnO4) and organic solvents (e.g. dimethylformamide or tetrahydrofuran), which make these top-down routes not environmentally benign.24,25
Recently ultrasonication as a new method for the production of graphene layers starting from graphite flakes or particles has aroused extensive interest. The reduction in size is achieved by cavitation, which refers to the formation, growth, and implosive collapse of bubbles in a liquid.26 The speed of sound in a typical liquid is 1000 to 1500 m s−1, and ultrasonic wavelengths vary from roughly 10 cm down to 100 μm over a frequency range of 20 kHz to 15 MHz, much larger than the molecular size scale.27 Exfoliation of defect-free graphene in a liquid phase was first demonstrated using non-aqueous conditions in dimethylformamide28 and in a number of organic solvents, such as N-methyl-2-pyrrolidone, dimethylacetamide, and ortho-dichlorobenzene.21 Due to the significant advantages of aqueous systems over non-aqueous systems (e.g., lower costs and fewer potential health risks and environmental issues), the use of water-based systems has attracted much attention in recent years. The ultrasonication-assisted exfoliation of graphite in an aqueous system using the cationic surfactant sodium dodecyl benzene sulfonate was first reported.23 Direct exfoliation of graphite via sonication has more recently been achieved using biopolymers in aqueous solutions of gelatin29 and gum arabic,30–32 and also exfoliation of inorganic graphene analogue, MoS2, in an acid aqueous solution of chitosan.33 The results arising from these few works make ultrasonication an extremely promising technique for large-scale production of good quality and cheap graphene. However, the potential of this technique is still far from fully understood.
The aim of this work is to evaluate the capability of polysaccharides in assisting the exfoliation of graphite into graphene nanosheets by using three different biopolymers (the positively charged chitosan, the neutral pullulan, and the anionic alginate) under ultrasonication conditions. The effects of the polysaccharide structure, initial graphite concentration, ultrasonication condition on the exfoliation efficiency and quality of the graphene nanosheets are investigated systematically.
Experimental
Reagents and chemicals
Chitosan (viscosity < 200 cP, degree of deacetylation: 85–95%) was purchased from Shanglong Aokang Bio Ltd., China. Pullulan (PF-20 grade, Mw ∼ 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) was obtained from Hayashibara Biochemical Laboratories Inc., Okayama, Japan. Sodium alginate (molecular weight 80000–120000; medium viscosity, viscosity of 2% solution in water at 25 °C ≥2000 cP; mannuronic/guluronic ratio of 1.56), graphite powder of purity of 99%, and acetic acid were supplied by Sigma-Aldrich, UK.
000 Da) was obtained from Hayashibara Biochemical Laboratories Inc., Okayama, Japan. Sodium alginate (molecular weight 80000–120000; medium viscosity, viscosity of 2% solution in water at 25 °C ≥2000 cP; mannuronic/guluronic ratio of 1.56), graphite powder of purity of 99%, and acetic acid were supplied by Sigma-Aldrich, UK.
Direct exfoliation of graphite
Pullulan and alginate (1.0 g) were separately dissolved in 20 ml of distilled water (DI), whereas chitosan (0.4 g) was dissolved in 20 ml of DI with 1 wt% acetic acid. Graphite powder was dispersed in the aqueous biopolymer solutions and treated using an ultrasonic processor UP200S (maximum power = 200 W, frequency = 24 kHz, Hielscher, Teltow, Germany) equipped with a cone frustum titanium sonotrode (model micro tip S3, tip diameter = 3 mm, maximum amplitude = 210 μm, acoustic power density or surface intensity = 460 W cm−2) under the following conditions: 0.5 cycle and 50% amplitude, for a period of 10, 20, 30, and 60 min, respectively. Subsequently, mixtures were centrifuged at 1500 rpm for 60 min to remove unexfoliated graphite particles and then washed 5 times and again centrifuged at 5000 rpm for 20 min to remove excess biopolymers. The resultant dark-gray solutions were vacuum-dried at 40 °C until no mass-loss. The resulting polymer–graphene powders were re-dispersed in water (1 mg ml−1 for pullulan and chitosan; 0.18 mg ml−1 for alginate) for characterization. Graphene sheets obtained by pullulan-, alginate-, and chitosan-assisted ultrasonication were indicated as pull-G, alg-G, and chit-G, respectively.Characterization
The yield of the overall process (i.e., the amount of exfoliated graphene sheets arising from the ultrasonication of graphite mediated by the polysaccharides) was determined by weighing in an analytical balance (Sartorius M-Power AZ214, Göttingen, Germany) the dried graphene arising from the centrifugations and washing cycles. The final concentration was expressed in mg ml−1 as a mean of three replicates. The same dried biopolymer–graphene samples were used to prepare a series of diluted dispersions, which allowed for the determination of the extinction coefficient using the Lambert–Beer law:| A = εlc | (1) |
The morphology and dimension of the graphene samples were observed by using transmission electron microscopy (TEM, JEOL 2000FX, 200 kV) and atomic force microscopy (AFM, Nanoscope V Multimode, Bruker, Germany). Raman spectroscopy measurements were recorded by a Renishaw inVia Raman spectrometer with an Ar-ion laser at an excitation wavelength of 514.5 nm, at ambient temperature. X-ray photoelectron spectroscopy (XPS) measurements were performed in an XM1000 instrument (Omicron NanoTechnology GmbH, Germany) equipped with a monochromatic Al Kα source. Data analysis was carried out using the CasaXPS package, using Shirley backgrounds, mixed Gaussian–Lorentzian (Voigt) line shapes, and asymmetrical parameters for the sp2 graphitic components. Thermogravimetric analysis (TGA) was carried out to estimate the amount of the residual biopolymer on the exfoliated graphene sheets after the 5 washing cycle, as described above. The analyses were run from 25 °C to 1000 °C at a linear heating rate of 10 °C min−1, using a TGA/DSC 2 instrument (Mettler Toledo, Switzerland) in an inert environment (50 ml min−1 N2). At least three replicates were used for each sample.
Results and discussion
Optimization of the exfoliation process
Graphene exfoliation was prepared in a pullulan aqueous solution under ultrasonication treatment. The efficiency of the graphene exfoliation was investigated by varying the initial graphite concentration, the concentration of pullulan in water, and the ultrasonication time. The optimized protocol was then extended to exfoliate graphite in chitosan- and alginate-solutions. As shown in Fig. 1, the final concentration of graphene in the pullulan solution scaled proportionally with the initial concentration of graphite. Therefore, the initial concentration of graphite was fixed at 10 mg ml−1. Using this concentration in a pullulan water solution of 10 mg ml−1, the amount of exfoliated graphene was about 0.54 mg ml−1 (see the half-square data point in the lower right of Fig. 1). For the same graphite concentration (10 mg ml−1) and the highest pullulan concentration of 50 mg ml−1, the amount of exfoliated graphene obtained was 2.3 mg ml−1 (see the half square data point in the upper left of Fig. 1). The final concentration of graphene in the pullulan solution scaled proportionally with the initial concentration of pullulan within the range 10–50 mg ml−1 (data not shown). We thus decided to set the pullulan concentration at 50 mg ml−1 (higher concentrations led to increasingly high viscosities).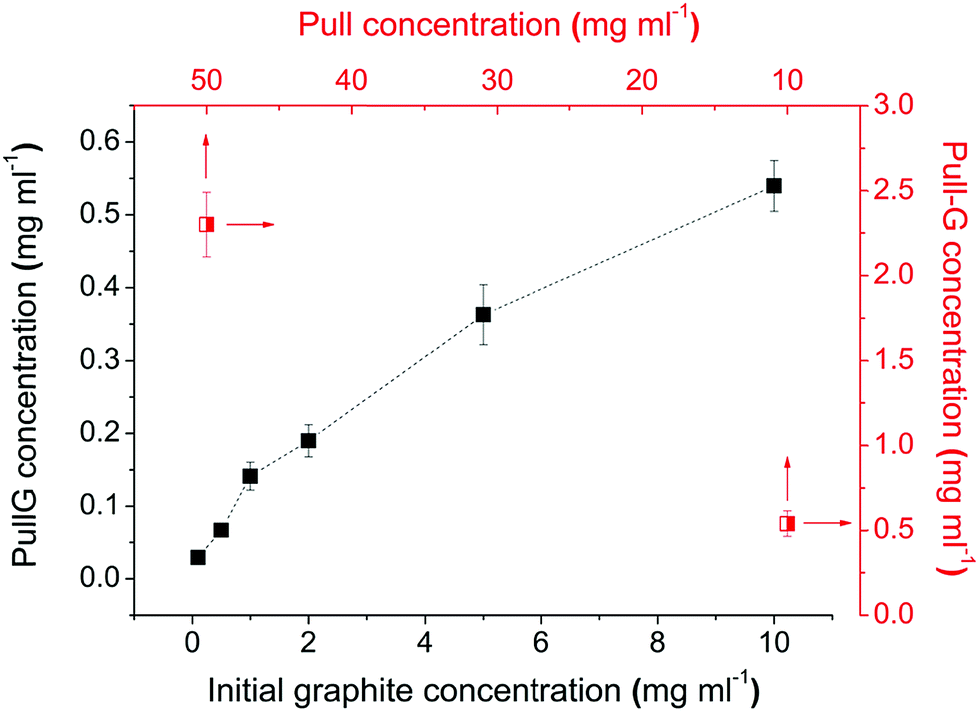 | ||
| Fig. 1 Effect of initial graphite concentration (full squared), and pullulan concentration (half squared) on the yield of graphene exfoliation (ultrasonication time 30 min). | ||
The effect of sonication time on the quality of graphene flakes in pullulan solution was observed by TEM. As shown in Fig. 2a–d, the pullulan-assisted ultrasonication process yielded thin and semi-transparent graphene flakes consisting of both individual and few stacked layers. After the observation of a reasonable number of graphene flakes for each sonication time, it was possible to consider the 30 minutes treatment a good compromise between number of layers, lateral dimensions, and surface area of the sheets (Fig. 2a–d).
 | ||
| Fig. 2 TEM images of pull-G for: (a) 10 min; (b) 20 min; (c) 30 min; (d) 60 min; (e) chit-G for 30 min; (f) alg-G for 30 min. | ||
Chitosan- and alginate-assisted ultrasonicated graphene sheets were thus obtained according to the optimized conditions for pullulan-assisted graphite exfoliation (i.e.: initial graphite concentration = 10 mg ml−1; polymer concentration = 50 mg ml−1 for alginate and 20 mg ml−1 for chitosan, the latter due to the high viscosity of the resulting water dispersion; sonication time = 30 min). The 30 minute sonication was more effective on chitosan than alginate, in terms of both number of stacked layers (visually detectable by the transparency of the flakes to the electron beam) and lateral dimensions (Fig. 2e and f). The yield of the process was 0.18 mg ml−1 for alg-G and 5.50 mg ml−1 for chit-G.
The successful exfoliation of graphite due to acoustic cavitation was further confirmed by visual inspection of the freshly-prepared water suspensions (Fig. 3), in line with previous works.29–32
 | ||
| Fig. 3 Digital images of freshly-prepared graphene (10 mg ml−1) in alginate (50 mg ml−1), chitosan (20 mg ml−1), and pullulan (50 mg ml−1) water dispersions after (a) 24 h; (b) 7 days; and (c) 6 months storage at room temperature. | ||
Adsorption of polysaccharides on the graphene surface
XPS and TGA analyses were carried out to characterize the adsorption of the polysaccharides on the graphene surfaces. As shown by the XPS survey spectra in Fig. 4, a dramatic increase in oxygen (peak at 531.9 eV) in pull-G, alg-G, and chit-G sheets and a new peak related to nitrogen (peak at 399.8 eV) in only chit-G sheets were observed, compared to pristine graphite, where a main peak at 284.4 eV (due to the presence of sp2 C–C bonds) was present. The atomic composition of the polysaccharide–graphene samples (Table 1) reveals that the highest amount of oxygen (16.03%) was measured on chit-G samples, which also had a significant presence of nitrogen (3%) due to –NH2 and –OH containing units.33 This further supports our previous observations on the preferential interaction of graphene with chitosan.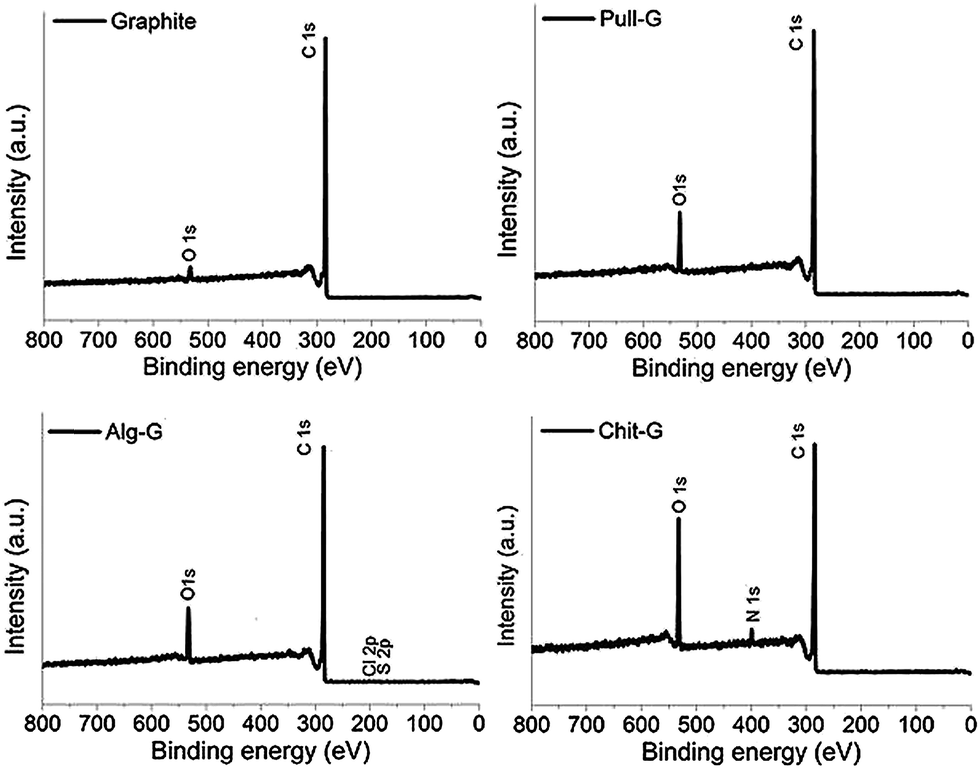 | ||
| Fig. 4 XPS survey spectra of pristine graphite powder, pull-G, alg-G, and chit-G. | ||
| Material | Total composition (%) | C:O ratio |
||||
|---|---|---|---|---|---|---|
| C | O | N | S | Cl | ||
| Graphite | 97.14 | 2.86 | — | — | — | 33.97 |
| Pull-G | 91.78 | 8.22 | — | — | — | 11.17 |
| Alg-G | 89.20 | 10.23 | — | 0.29 | 0.28 | 8.72 |
| Chit-G | 80.97 | 16.03 | 3.00 | — | — | 5.05 |
As determined by TGA (Fig. S1†), the amount of pullulan, alginate, and chitosan polymers adsorbed on the graphene surface was estimated to be approximately ∼2.5 wt%, ∼1.5 wt%, and ∼8.5 wt%, respectively (see the insets of Fig. S1†). These values are much lower if compared to gum arabic–graphene sheets, for which the residual biopolymer amount was 23% at 550 °C,30 48.7% at 750 °C,31 and 56% at 800 °C.32 However, it should be noted here that the amount of biopolymer found on pull-G samples before the 5 washing steps ranged between 48% and 68%, in line with previous results on gum arabic–graphene bionanocomposites.
These results confirm the preferential affinity of graphene for the three biopolymers according to the following decreasing order: chitosan > pullulan > alginate. Noteworthy, residual chitosan (8.5 wt%) and pullulan (2.5 wt%) made possible the re-dispersibility of graphene particles in water (Fig. 5). In particular, while chitosan provided stable homogeneous dispersions of graphene in water without reaggregation being observed, pull-G dispersions displayed precipitation after storage at room temperature for one week. Alg-G dispersions exhibited the same behavior observed for the parental polymer solution, i.e. the biopolymer–graphene powder redispersed in water was stable for only 1 day, after which reaggregation and precipitation occurred. Although only few studies took into account the redispersibility degree of exfoliated graphene in aqueous media,31,32,34 this is an important aspect to be considered in liquid exfoliation systems, because it may allow fulfilling specific requirements for diverse applications of exfoliated graphene. For example, the adsorbed pullulan and chitosan would be of great advantage in the preparation of water-based graphene bionanocomposites according to the “one-pot” procedure,29,35–38 because the affinity between polymer and filler is inherently achieved. This would represent the simplest, most efficient, and most environmentally friendly strategy for the preparation of graphene-based bionanocomposite materials.37
 | ||
| Fig. 5 Digital images of redispersed alg-G (0.18 mg ml−1), chit-G (1 mg ml−1), and pull-G (1 mg ml−1) powders in water after (a) 24 h and (b) 7 days storage at room temperature. | ||
We acquired the same kind of information from the extinction coefficient values of a series of diluted water dispersions prepared for each biopolymer–graphene system after the 5-step washing procedure. The absorbance unit values for the 3 systems increased monotonically with the concentration (Fig. S2†). The extrapolated extinction coefficients (ε) were 525 ml mg−1 m−1, 1240 ml mg−1 m−1, and 2287 ml mg−1 m−1 for alg-G, pull-G, and chit-G, respectively. As explained by Su and co-workers,39 the different extinction coefficient calculated for the three systems can be explained by considering the light absorption characteristics of mono- or multi-layered graphene, which depend on lateral size distribution, number of layers per flake, and number and type of functional groups.
In particular, a higher content of small flakes and fewer layers per flake concurrently give smaller extinction coefficients, which were ascribed to the shrinkage of the π-conjugated system at 660 nm. By contrast, high extinction coefficients are encountered for high contents of functional groups because of the increase in the auxochromic effect. Therefore, the higher extinction coefficient calculated for the chitosan-coated graphene flakes, compared to both pullulan– and alginate–graphene system, can be attributed to both a “surface chemistry” effect (amino groups, hydroxyl groups, and acetyl groups along its backbone) and a “mass” effect, being the amount of chitosan per unit area adsorbed on the graphene flakes thicker compared to pullulan and—especially—alginate.
Morphology and quality of exfoliated graphene sheets
We gathered information on both morphology and thickness of pull-G, alg-G, and chit-G nanosheets through AFM analyses. Representative AFM images are shown in Fig. 6. Graphene sheets with irregular edges can be clearly detected in the pull-G sample (Fig. 6a), whereas the alg-G sample apparently showed macroscopic aggregates (Fig. 6b). Chit-G AFM images exhibited a peculiar pattern, with graphene sheets noticeably masked by the polymer matrix (Fig. 6c). | ||
| Fig. 6 AFM height images (5 × 5 μm2) of (a) pull-G, (b) alg-G and (c) chit-G nanosheets deposited on mica substrates. | ||
As a general trend, the ultrasonication process mediated by pullulan yielded graphene sheets with a surface area mostly centered between 2 × 105 nm2 and 4.0 × 105 nm2, with a thickness between 0.7 nm and 1.5 nm. The surface area and the thickness of alg-G samples increased to approximately 5.7 × 105 nm2 and 1.3–5.5 nm, respectively, confirming the previous indication of the tendency of graphene sheets to restack. As for the chit-G samples, the surface area of the graphene sheets was approximately 1.8 × 105 nm2, while the thickness dramatically increased to 10–15 nm, which can be attributed to the large amount of polymer adsorbed on the graphene sheets. The presence of residual chitosan can also be inferred from the relatively high roughness (RMS = 3.7 nm on 2.5 × 2.5 μm2 area), whereas the remaining pullulan polymer in pull-G samples is unnoticed because of the even surface associated with very low roughness values.40 Arising from these observations, considering that the actual thickness of an individual graphene sheets is ∼0.34 nm,41 and in light of the residual polysaccharides adsorbed on the graphene sheets (∼2.5%, ∼1.5%, and ∼8.5% for pull-G, alg-G, and chit-G, respectively), the pull-G sheets are single or few layers (<5 layers); alg-G are few layers or quite thick sheets; and chit-G are single or few layers with a high amount of polymer adsorbed.
Raman spectrum of pristine graphite shows a dominant G band at 1582 cm−1 and two additional bands, D and 2D bands, located at 1350 cm−1 and 2700 cm−1, respectively (Fig. 7a), in line with the literature.42,43 After exfoliation, the D-band with a relatively strong intensity can be mainly ascribed to the increased fraction of graphene edges.43–45 Moreover, the intensity ratio of the D and G bands (ID/IG) reflects the structural defects and the indication of disorder.42 In this work, although the D-peak was present in all three exfoliated graphene samples (Fig. 7), the ID/IG ratio was reasonably low, as it increased from 0.08 for graphite to 0.18, 0.33, and 0.49 for alg-G, pull-G and chit-G, respectively (Fig. 7a), in line with the values found for gum arabic-assisted graphene sheets (0.29 and 0.25).30,32 This indicates that low-edge defects rather than basal plane defects arose from the sonication process,46 supporting the unaltered graphitic character of the flake basal planes after ultrasonication.30 Our results thus confirm that graphene sheets produced using polysaccharide-assisted ultrasound exfoliation are relatively defect-free30,32 compared to reduced GO, which is primarily due to the use of harsh oxidizing reagents used during the oxidation of graphene—the successive reducing process does not allow full recovery of the original graphitic structure.30
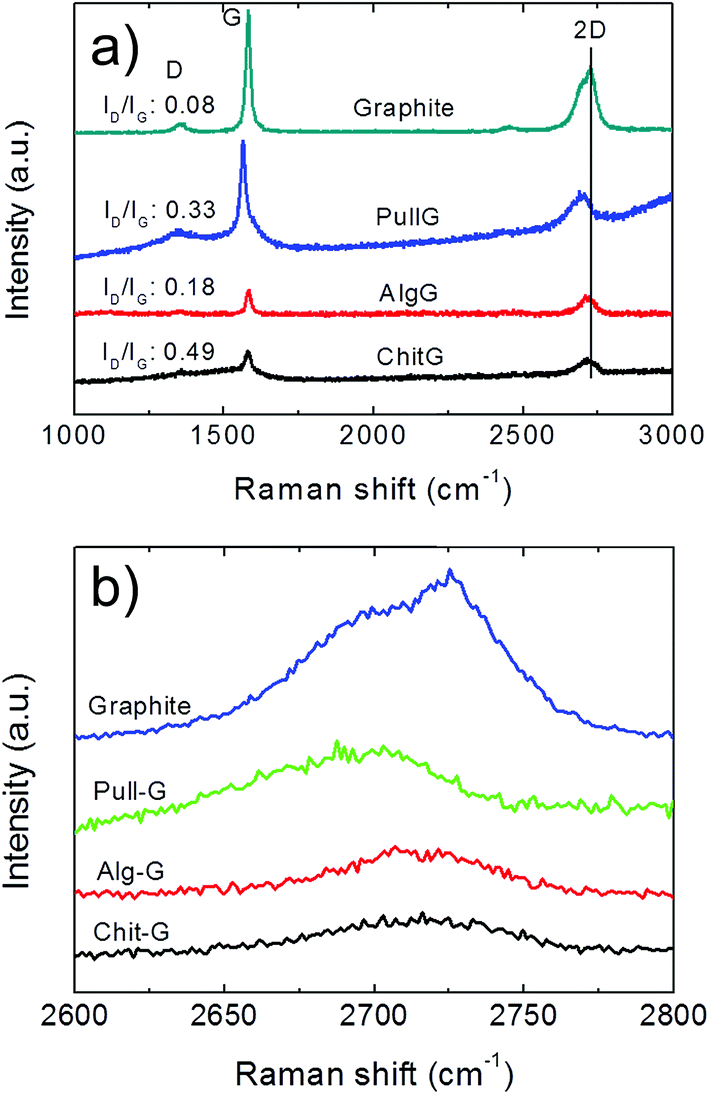 | ||
| Fig. 7 (a) Raman spectra of graphite, pull-G, alg-G, and chit-G; (b) detail of the 2D Raman band. | ||
The (I2D/IG) ratio and the full width at half maximum (FWHM) of the 2D band provide information on the average thickness of graphene sheets.47,48 In particular, the I2D/IG ratio decreased from ∼2.1 for single-layer graphene to ∼0.8 for quadruple-layer samples.48 We also found that FWHM is ∼30–35 cm−1 for individual graphene layers, and this value nearly doubles in all the three systems (53.34 cm−1, 54.72 cm−1, and 70.99 cm−1 for pull-G, alg-G, and chit-G, respectively), which is a clear indication of two layers43 and quadruple-layer samples.48
The shape of the 2D peak is another important parameter (Fig. 7b). The 2D peak of graphite spectrum consists of two components, 2D1 and 2D2. These two peaks disappear in a single graphene layer, which exhibits a single, sharp and intense 2D peak at lower wavenumbers, roughly four times more intense than the G peak. Bi-layer graphene has much a wider peak compared to single-layer graphene, whereas the peak of more than five layers graphene becomes hardly distinguishable from that of graphite due to similarity of the 2D-band in shape.42,45 Significant changes occurred in the shape of the 2D peak of the polysaccharides–graphene nanosheets compared to the pristine graphite powder, especially in terms of shifting toward lower wavenumbers (Fig. 7b).
These results confirmed that the polysaccharide-assisted ultrasonication of graphite powder was able to provide exfoliated graphene, most likely consisting of a mixture of mono-layer, bi-layer, and few-layer sheets—in any case, less than 5 layers—in particular for the pullulan-assisted ultrasonication, in agreement with the AFM results. The highest value of both ID/IG and FWHM for chitosan could be affected by the high amount of residual chitosan adsorbed on the surface of graphene, as discussed earlier.
Influence of the polysaccharide type on the stability of the ultrasonicated graphene water dispersions
One of the main findings arising from this study was the different behavior of the three polysaccharides to promote a long-lasting exfoliation of graphene sheets in aqueous medium. Indeed, the exfoliated flakes in chitosan and pullulan solutions were stable for long periods (over 6 months), unlike the alginate-based water suspension, which started to collapse after 24 h. A plausible explanation for our observations can tentatively be made in terms of thermodynamic compatibility between biopolymer and graphene.The preferential interaction between graphene and the polycationic chitosan can be due to the affinity between the non-polar chain segments of chitosan and the surface of graphene. This is corroborated by the high dispersive (apolar) component of the surface free energy measured for chitosan (∼47 mJ m−2),49 very close to that of graphene (46.7 mJ m−2),50 which would give a reason for the adsorption of chitosan molecules onto the surface of graphene through hydrophobic–hydrophobic interactions.35 The electrostatic repulsion between positively charged amino groups exposed to the aqueous medium would instead prevent the restacking and agglomeration of the exfoliated flakes, thus leading to a stable dispersion, as also confirmed by the zeta potential value measured for the freshly prepared chit-G water dispersion (ζ = 43.2 ± 0.5 eV, pH = 3.5).
Analogously, pullulan-based dispersions were stable for a long time due to the affinity between the biopolymer and the surface of graphene. Although highly polar and yet largely hydrophilic in nature, this exopolysaccharide exhibits quite a high dispersive component (∼44 mJ m−2),49 which would ensure the adsorption on the surface of the graphene sheets. The final colloidal stability in water is eventually achieved by the high affinity with the surrounding aqueous medium as well as by steric and/or depletion stabilization,51 although the lack of charged functional groups does not provide any electrostatic repulsion as seen for chitosan (in fact, the pull-G dispersions started to precipitate earlier than those based on chitosan). This was confirmed by the low zeta potential value of the freshly prepared pull-G water dispersion (ζ = 2.2 ± 0.2 eV, pH = 5).
Finally, the completely different behavior observed for the alginate dispersions can be again explained in terms of the affinity between the biopolymer and the graphene sheets. The very high zeta potential value of the freshly prepared alg-G water dispersion (ζ = −82.4 ± 0.7 eV, pH = 5) explained the initial excellent dispersibility of graphene in the alginate solution. However, it is plausible that the compatibility at the interface between the two entities (alginate and graphene) is thermodynamically unfavorable, as suggested by the dispersive component of the surface free energy measured for alginate (∼20 mJ m−2),52 far lower than for chitosan and pullulan. So the adsorption of the biopolymer on the graphene surface is somehow hindered, which would explain the restacking and precipitation of the graphene layers after ∼24 hours.
Eventually, all the above considerations seem to confirm that good compatibility (e.g., solvents, surfactants, polymers) for graphite are characterized by surface tensions in the region of 40–50 mJ m−2.21
Efficiency of the ultrasonication process
It is important to comment on the efficacy and efficiency of the ultrasonication process, especially in light of previous studies using high-intensity (interchangeably called “tip” or “sonotrode”) or low-intensity (e.g., ultrasound bath) ultrasonication methods. Table 2 summarizes the most relevant results reported in the literature on the ultrasound-assisted exfoliation of graphite in polymer–water mixtures. At first glance, our procedure involving chitosan as the ultrasonication-assisting biopolymer led to the highest yield reached so far (5.50 mg ml−1). Of course, this cannot be taken as an absolute value, as many parameters differ from one study to another (for example the sonication time may vary from 30 minutes to 100 hours). However, this work has demonstrated that pinpointing the best factors combination is of utmost importance to optimize the final yield and define a reproducible protocol for the liquid-phase exfoliation process.| Polymer type | CGmax (mg ml−1) | Sonication type | Sonication time (h) | CP (mg ml−1) | CGi (mg ml−1) | ε (ml mg−1 m−1) | Ref. |
|---|---|---|---|---|---|---|---|
| a CGmax: maximum achieved graphene concentration; CP: polymer concentartion; CGi: initial graphene concentration; ε = extinction coefficient; GMA–gum arabic: glycidyl methacrylate-functionalized gum arabic; PVP: polyvinylpyrrolidone. | |||||||
| Pullulan | 2.30 | Tip | 0.5 | 50 | 10 | 1240 | Present study |
| Alginate | 0.18 | Tip | 0.5 | 50 | 10 | 525 | Present study |
| Chitosan | 5.50 | Tip | 0.5 | 20 | 10 | 2287 | Present study |
| GMA–Gum arabic | 1.12 | Bath | 5 | 30 | 80 | 1390 | 31 |
| Gum arabic | 0.69 | Bath | 8 | 140 | 10 | 1390 | 32 |
| Gelatin | 0.60 | Bath | 8 | 20 | 200 | 1390 | 29 |
| Pyrene-functionalised block copolymer | 0.39 | Bath | 6 | 20 | 0.5 | — | 36 |
| Acrylate polymer/ethanol | 4.00 | Bath | 24 | 20 | 200 | 2607 | 55 |
| Gum arabic | 0.60 | Bath | 100 | 50 | 10 | 5422 | 30 |
| PVP | 0.42 | Tip | 1 | 20 | 40 | 1293 | 34 |
| PVP | 0.10 | Bath | 9 | 20 | 5 | — | 51 |
Equally important is the type of ultrasound wave used to promote the exfoliation. Indisputably, high-intensity ultrasound is far more effective than low-intensity ultrasound waves, such as those generated by the ultrasound bath, basically because of the greatly higher energy input involved, especially at local levels. This is the reason for the 430 h needed to disperse graphene by bath sonication in both non-aqueous N-methyl-pyrrolidone53 and sodium cholate water-based solutions,54 which yielded at most 1.2 mg ml−1 and 0.3 mg ml−1 graphene concentration, respectively. A higher yield value (4 mg ml−1) by bath sonication was achieved after 24 hours sonication of graphite in ethanol assisted by an acrylate polymer.55 We found only one work using a tip sonicator to exfoliate graphite in an aqueous medium containing PVP as a non-ionic polymer. However, after 1 h sonication, the yield was 0.42 mg ml−1 of single- and few-layer graphene,34 and about 0.10 mg ml−1 single-layer graphene was obtained after 9 h bath sonication using the same polymer.51
More recently, Guardia and co-workers noticed a remarkable increase of the amount of exfoliated graphene analogues, MoS2 and WS2, with increasing the power intensity.56 In our work, we used a power of 16.25 W for 30 minutes of sonication, with energy consumption (energy output per unit volume) of 731 W s ml−1. While high energy inputs may provide higher exfoliation, smaller lateral dimensions of the graphene sheets can be a concomitant undesired side effect.56 In addition, prolonged sonication times, instead of providing additional benefit in yields, can lead to more intense damage of the graphene lattice21 and higher and worthless energy input, as already demonstrated for the ultrasound-assisted exfoliation of clays.40 Therefore, to make ultrasonication an effective and efficient approach for mass production of high-quality graphene sheets, a thoughtful balance between yield, quality of the graphene sheets, and overall costs involved is necessary.
Conclusions
In this work we investigated the capability of non-ionic pullulan, anionic alginate, and cationic chitosan to assist the ultrasonication-mediated exfoliation of graphite into graphene nanosheets in an aqueous medium. Out of the three systems, pullulan and chitosan were more effective in exfoliation of graphite than alginate. This method yielded exfoliated mono-, bi-, and few-layer graphene sheets with only low lateral (edges) defects. The adsorption of biopolymers on graphene surface affords a long-lasting stability (more than 6 months) of the aqueous dispersion.Besides allowing new potential uses of biomass resources (e.g., chitin and algae), the proposed protocol represents a high-throughput, high-yield, economical, and scalable route for new applications of graphene for packaging applications (e.g., food packaging), where the intrinsic properties of graphene such as high elastic modulus and gas barrier properties are sought after.
References
- S. Sheshmani and R. Amini, Carbohydr. Polym., 2013, 95, 348 CrossRef CAS PubMed.
- L. Imperiali, C. Clasen, J. Fransaer, C. W. Macosko and J. Vermant, Mater. Horiz., 2014, 1, 139 RSC.
- I. Uysal Unalan, G. Cerri, E. Marcuzzo, C. A. Cozzolino and S. Farris, RSC Adv., 2014, 4, 29393 RSC.
- A. Ashori, Polym. Eng. Sci., 2013, 54, 2258 Search PubMed.
- D. Lee, M.-C. Choi and C.-S. Ha, J. Polym. Sci., A: Polym. Chem., 2012, 50, 1611 CrossRef CAS.
- Y. Lee, D. Kim, J. Seo, H. Han and S. B. Khan, Polym. Int., 2013, 62, 1386 CrossRef CAS.
- A. Al-Jabareen, H. Al-Bustami, H. Harel and G. Marom, J. Appl. Polym. Sci., 2013, 128, 1534 CAS.
- D. Kim, H. Kwon and J. Seo, Polym. Compos., 2014, 35, 644 CrossRef CAS.
- O. C. Compton, S. Kim, C. Pierre, J. M. Torkelson and S. T. Nguyen, Adv. Mater., 2010, 22, 4759 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- W. Chen, L. Yan and P. R. Bangal, J. Phys. Chem. C, 2010, 114, 19885 CAS.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2009, 4, 25 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS PubMed.
- H.-B. Zhang, J.-W. Wang, Q. Yan, W.-G. Zheng, C. Chen and Z.-Z. Yu, J. Mater. Chem., 2011, 21, 5392 RSC.
- O. Akhavan, Carbon, 2010, 48, 509 CrossRef CAS PubMed.
- W. Lv, D.-M. Tang, Y.-B. He, C.-H. You, Z.-Q. Shi, X.-C. Chen, C.-M. Chen, P.-X. Hou, C. Liu and Q.-H. Yang, ACS Nano, 2009, 3, 3730 CrossRef CAS PubMed.
- O. Akhavan, Carbon, 2011, 49, 11 CrossRef CAS PubMed.
- B. Li, X. Zhang, X. Li, L. Wang, R. Han, B. Liu, W. Zheng, X. Li and Y. Liu, Chem. Commun., 2010, 46, 3499 RSC.
- W. Zhao, M. Fang, F. Wu, H. Wu, L. Wang and G. Chen, J. Mater. Chem., 2010, 20, 5817 RSC.
- J. Chen, M. Duan and G. Chen, J. Mater. Chem., 2012, 22, 19625 RSC.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS PubMed.
- H. Bai, C. Li and G. Shi, Adv. Mater., 2011, 23, 1089 CrossRef CAS PubMed.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217 CrossRef CAS PubMed.
- S. Vadukumpully, J. Paul and S. Valiyaveettil, Carbon, 2009, 47, 3288 CrossRef CAS PubMed.
- T. Hielscher, Proceeding of European Nanosystems Conference ENS '05, Paris, France, 2005 Search PubMed.
- H. Xu, B. W. Zeiger and K. S. Suslick, Chem. Soc. Rev., 2013, 42, 2555 RSC.
- P. Blake, P. D. Brimicombe, R. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson, E. W. Hill, A. K. Geim and K. S. Novoselov, Nano Lett., 2008, 8, 1704 CrossRef PubMed.
- Y. Ge, J. Wang, Z. Shi and J. Yin, J. Mater. Chem., 2012, 22, 17619 RSC.
- V. Chabot, B. Kim, B. Sloper, C. Tzoganakis and A. Yu, Sci. Rep., 2013, 3, 1378 Search PubMed.
- J. Fan, Z. Shi, J. Wang and J. Yin, Polymer, 2013, 54, 3921 CrossRef CAS PubMed.
- J. Fan, Z. Shi, Y. Ge, J. Wang, Y. Wang and J. Yin, J. Mater. Chem., 2012, 22, 13764 RSC.
- M. Fang, J. Long, W. Zhao, L. Wang and G. Chen, Langmuir, 2010, 26, 16771 CrossRef CAS PubMed.
- A. S. Wajid, S. Das, F. Irin, H. S. T. Ahmed, J. L. Shelburne, D. Parviz, R. J. Fullerton, A. F. Jankowski, R. C. Hedden and M. J. Green, Carbon, 2012, 50, 526 CrossRef CAS PubMed.
- X. Feng, X. Wang, W. Xing, K. Zhou, L. Song and Y. Hu, Compos. Sci. Technol., 2014, 93, 76 CrossRef CAS PubMed.
- Z. Liu, J. Liu, L. Cui, R. Wang, X. Luo, C. J. Barrow and W. Yang, Carbon, 2013, 51, 148 CrossRef CAS PubMed.
- D. Nuvoli, M. Rassu, V. Alzari, R. Sanna, G. Malucelli and A. Mariani, Compos. Sci. Technol., 2014, 96, 97 CrossRef CAS PubMed.
- X. Zheng, Q. Xu, J. Li, L. Li and J. Wei, RSC Adv., 2012, 2, 10632 RSC.
- R. Su, S. F. Lin, D. Q. Chen and G. H. Chen, J. Phys. Chem. C, 2014, 118, 12520 CAS.
- L. Introzzi, T. O. J. Blomfeldt, S. Trabattoni, S. Tavazzi, N. Santo, A. Schiraldi, L. Piergiovanni and S. Farris, Langmuir, 2012, 28, 11206 CrossRef CAS PubMed.
- P. Nemes-Incze, Z. Osváth, K. Kamarás and L. P. Biró, Carbon, 2008, 46, 1435 CrossRef CAS PubMed.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47 CrossRef CAS PubMed.
- D. Graf, F. Molitor, K. Ensslin, C. Stampfer, A. Jungen, C. Hierold and L. Wirtz, Nano Lett., 2007, 7, 238 CrossRef CAS PubMed.
- C. Casiraghi, A. Hartschuh, H. Qian, S. Piscanec, C. Georgi, A. Fasoli, K. S. Novoselov, D. M. Basko and A. C. Ferrari, Nano Lett., 2009, 9, 1433 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- S. Gayathri, P. Jayabal, M. Kottaisamy and V. Ramakrishnan, AIP Adv., 2014, 4, 027116 CrossRef PubMed.
- D. Graf, F. Molitor, K. Ensslin, C. Stampfer, A. Jungen, C. Hierold and L. Wirtz, Solid State Commun., 2007, 143, 44 CrossRef CAS PubMed.
- A. A. Green and M. C. Hersam, Nano Lett., 2009, 9, 4031 CrossRef CAS PubMed.
- S. Farris, L. Introzzi, P. Biagioni, T. Holz, A. Schiraldi and L. Piergiovanni, Langmuir, 2011, 27, 7563 CrossRef CAS PubMed.
- S. Wang, Y. Zhang, N. Abidi and L. Cabrales, Langmuir, 2009, 25, 11078 CrossRef CAS PubMed.
- A. B. Bourlinos, V. Georgakilas, R. Zboril, T. A. Steriotis, A. K. Stubos and C. Trapalis, Solid State Commun., 2009, 149, 2172 CrossRef CAS PubMed.
- T. Çaykara, S. Demirci, M. S. Eroğlu and O. Güven, Polymer, 2005, 46, 10750 CrossRef PubMed.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864 CrossRef CAS PubMed.
- M. Lotya, P. J. King, U. Khan, S. De and J. N. Coleman, ACS Nano, 2010, 4, 3155 CrossRef CAS PubMed.
- Z. Sun, S. Pöller, X. Huang, D. Guschin, C. Taetz, P. Ebbinghaus, J. Masa, A. Erbe, A. Kilzer, W. Schuhmann and M. Muhler, Carbon, 2013, 64, 288 CrossRef CAS PubMed.
- L. Guardia, J. I. Paredes, R. Rozada, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, RSC Adv., 2014, 4, 14115 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: TGA traces of graphite, pullulan, alginate, chitosan, and pullulan–graphene, alginate–graphene and chitosan–graphene exposed to a N2 atmosphere (Fig. S1); absorbance unit values for different concentrations of pullulan–graphene, chitosan–graphene, and alginate–graphene water dispersions (Fig. S2). See DOI: 10.1039/c4ra16947f |
| This journal is © The Royal Society of Chemistry 2015 |