
Photophysics of tungsten–benzylidyne complexes derived from s-indacene: synthesis, characterization and DFT studies†
Abstract
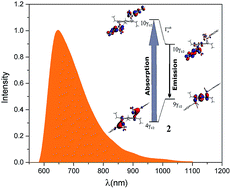
The photophysics of the mono- and homobimetallic complexes of tungsten–benzylidyne derived from s-indacene have been examined by using absorption and emission. Theoretical calculations of these compounds were carried out to gain further understanding of these novel molecular systems. Consistent with this prediction, each of the complexes displays a weak, mid-visible absorption band which is attributed to the d → π* transition. The tungsten complexes also exhibit luminescence with a lifetime in the 5–6 ns regime.
 Please wait while we load your content...
Please wait while we load your content...

