
Design of protein crystals in the development of solid biomaterials
Satoshi Abe and
Takafumi Ueno*
Department of Biomolecular Engineering, Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, 4259-B55, Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan. E-mail: tueno@bio.titech.ac.jp
First published on 18th February 2015
Abstract
The design of protein crystals has become a growing field in materials science and biotechnology. Protein crystals have immense potential for use as new porous materials that allow the precise arrangement of exogenous compounds by metal coordination and chemical conjugation in the solvent channels of the crystals. Generation of novel protein crystal lattices and regulation of chemical environments by design of protein interfaces represent important efforts in this field. In this review, we focus on recent advances in the preparation of 2D and 3D protein crystals by several approaches including protein engineering, ligand-mediated assembly, interfacial metal coordination, symmetric assembly by engineered proteins, genetic fusion of protein domains, and computational design of protein interfaces. A major advantage of using protein crystals as solid materials is that they can be utilized as porous biomaterials for accumulation of functional molecules, such as metal ions, metal complexes and inorganic nanoparticles. Protein crystals can be used in applications such as separation processes, heterogeneous enzymatic catalysis and drug delivery systems. It is expected that engineering of protein crystals will provide remarkable advances in the field of materials science.
1. Introduction
The design of protein assemblies has been the subject of significant interest in the field of bionanotechnology and biomaterial sciences. Protein assemblies with highly ordered nanostructures can be utilized as reaction vessels and molecular templates in efforts to create new functions.1–4 Various protein assemblies can provide specialized chemical environments which can be harnessed for incorporation and/or display of inorganic materials and metal complexes as well as functional organic molecules with catalytic, magnetic and optical properties. Typical protein assemblies suitable for such applications are shown in Fig. 1. Protein cages provided by structures such as viruses and ferritin, have been investigated as molecular templates for preparation of metallic nanoparticles and immobilization of metal complexes within the cage in efforts to produce specialized systems for catalysis and drug delivery (Fig. 1a).5–10 Various functional molecules such as organic chromophores, metal complexes, and proteins, which are precisely aligned on the surface of the protein tubes, have been used in the construction of systems for light-harvesting, catalysis, and in vivo imaging (Fig. 1b).11–13 Therefore, the development of advanced materials by construction of artificial protein assemblies has been accelerating as reported in recent reviews.14–17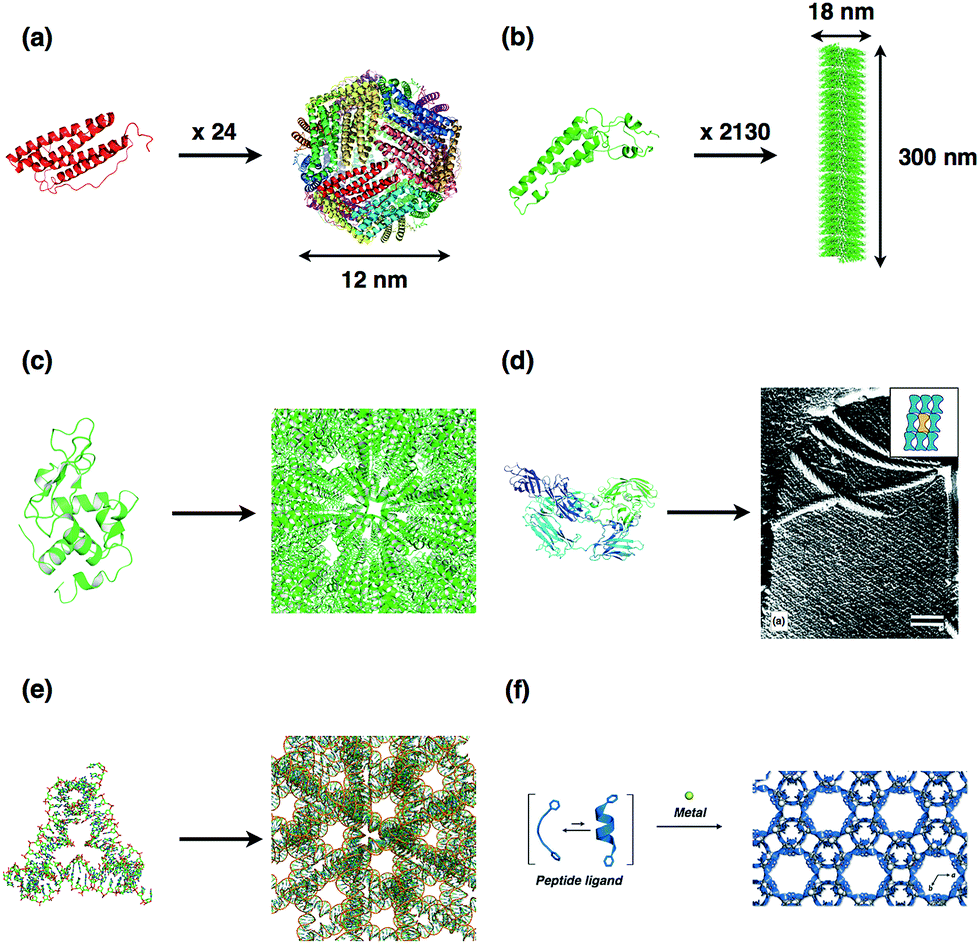 | ||
| Fig. 1 Typical biomolecular assembly structures; (a) ferritin, (b) tobacco mosaic virus (TMV), (c) lysozyme crystal, (d) S-layer protein and assembly structure on the cell surface, (e) DNA crystal, and (f) metal induced peptide crystal. The structures of (a), (b), (c), (d), and (e) were obtained from PDB codes 1DAT, 1VTM, 193L, 4AQ1, and 3GBI, respectively. Adapted from ref. 20 and 27 with permission for (d) and (f) from Elsevier Ltd. and Wiley-VCH, respectively. | ||
Recently developed protein assembly engineering technologies have been applied to protein crystals in efforts to advance the field of materials science. Protein crystals have highly ordered 2D and 3D arrangements of protein molecules in the solid state with infinite assembling structures (Fig. 1c and d).18–23
Margolin et al. have reported that protein crystals typically comprise solvent channels ranging in size from 30% to 65% of the total crystal volume with high porosity (0.5–0.8) and a wide range of pore sizes (20–100 Å).18,19 One of the advantages of using crystals as molecular templates is that they can be utilized as porous biomaterials for applications in separation processes, heterogeneous enzymatic catalysis and drug delivery.18 Moreover, exogenous compounds such as metal ions, metal complexes and inorganic nanoparticles are accumulated in an orderly fashion in the solvent channels. This is an important characteristic for development of functions such as catalysis, magnetism and optics.3,4,24 The size and morphology of the pores of protein crystals depends on the surface characteristics of proteins and the crystallization conditions (Fig. 1c). Recently, 3D crystals of other biomolecules such as DNA and peptides have also been reported. 3D crystals of DNA have been designed as solid materials for encapsulation of enzymes into solvent channels (Fig. 1e).25,26 Metal–peptide frameworks have been designed by metal coordination with short peptides for applications of chiral recognition of chiral molecules (Fig. 1f).27,28 These examples suggest that crystalline assemblies of biomacromolecules are attractive candidates for development of novel solid materials in nanotechnology and nanomaterial sciences. In this review, we focus on recent developments in the construction of protein crystals with new lattice structures by protein engineering, and in functionalization of solvent channels of protein crystals by accumulation of metal ions and metal complexes.
2. Design for protein crystallization
Protein crystallization proceeds in two steps: nucleation and growth. Nucleation process is the most important and difficult to obtain the protein crystals. In nucleation process of crystals, the formation of partially ordered protein assemblies or paracrystalline intermediates is occurred as critical nuclei. The growth of protein crystals mainly proceeds by dislocation growth and two-dimensional nucleation of proteins. It is important to promote the ordered association of protein monomers, which avoid the random aggregation and precipitates for formation of nuclei.29 Several approaches have been developed to design and form nuclei of protein crystals with novel lattice structures.30–38 In general, symmetrical proteins such as homodimers nucleate more easily than asymmetric proteins.39 Therefore, design of protein interfaces for formation of symmetric assemblies represents an important effort in preparation of artificial protein crystals. Protein assemblies with defined structures have been recently constructed by rational design of interfaces between protein monomers.14–17 These strategies can be expanded to prepare 2D and 3D infinite crystalline arrays of proteins. Here, we describe several approaches in the synthesis of 2D and 3D crystalline arrays: (i) ligand-mediated assembly,30,31 (ii) interfacial metal coordination,32–34,40,41 (iii) generation of symmetry by design of variants,35,36 (iv) genetic fusion of two protein domains which naturally form oligomeric proteins,37 and (v) computational design of protein interfaces (Fig. 2).38 | ||
| Fig. 2 Protein interfacial design to form crystalline structures. (a) Ligand-mediated crystallization of carbohydrate binding proteins with biligand. (b) 2D or 3D crystals mediated by metal coordination of hemoproteins. (c) Disulfide bonding between protein monomers for oligomerization of synthetic protein assemblies. (d) 2D crystallization by self-assembly of fusion proteins, which are natural oligomer assemblies. | ||
2.1. Ligand-mediated crystallization
Small ligands, which are interacted with proteins, are utilized for assembling the proteins to form crystals. Freeman and co-workers have designed three-dimensional diamond-like lattice structures based on the assembly of the tetrahedral lectin concanavalin A (carbohydrate binding proteins, ConA) and a biligand of mannose derivatives.30 ConA has four binding sites for α-D-mannopyranoside or α-D-glucopyranoside. When two equivalents of bismannopyranoside are reacted with ConA, three-dimensional crystalline protein precipitants are formed through binding of two-headed carbohydrates to tetrameric concanavalin (Fig. 3). Electron transmission micrographs of the crystalline precipitants show that protein particles are highly ordered with distances of 6.9 ± 0.3 nm between the centers of neighboring molecules. The X-ray diffraction patterns of the crystalline precipitants at 6 Å revealed a pseudo-cubic orthorhombic unit cell (space group F222). This technology of ligand-mediated assembly into crystals could be applied in further engineering of three-dimensional protein crystals.31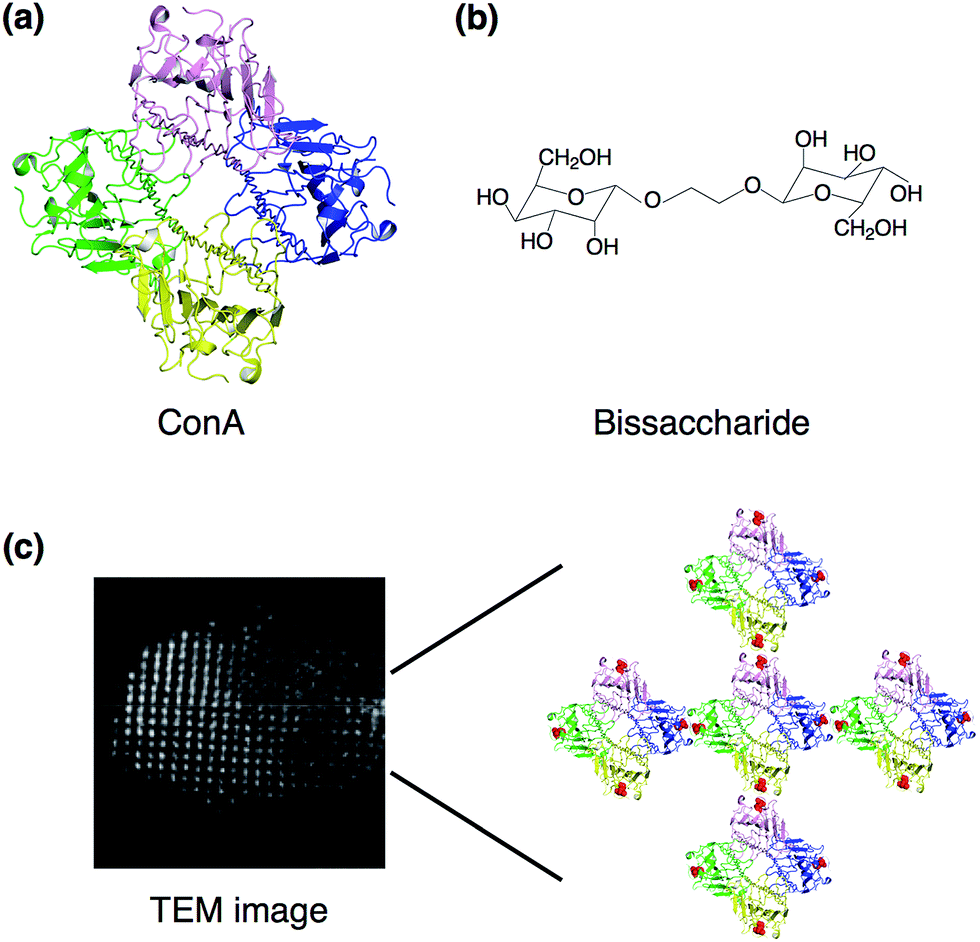 | ||
| Fig. 3 Ligand-mediated crystallization. (a) Crystal structure of tetrameric lectin concanavalin A; structure taken from a PDB code 5CNA. (b) Structure of bismannopyranoside. (c) TEM image of negatively stained crystal and assembly of tetrameric ConA with bisaccharide. Adapted from ref. 29 with permission for (b) and (c) from Wiley-VCH. | ||
Recently, an inter-penetrating structure of a protein crystal framework has been prepared using a similar lectin-carbohydrate interaction in combination with dimerization of Rhodamine B.31
2.2. Crystallization mediated by metal coordination
Metal coordination with amino acid residues on protein surface facilitates crystallization of proteins by forming symmetrical assemblies. In 1991, Artymiuk et al. reported the crystallization of the H-type of ferritin (H-ferritin), an iron storage spherical caged protein with an 8 nm inner diameter, which was facilitated by surface engineering.32 Although L-ferritin can be crystallized in the presence of Cd2+ ions, H-ferritin is not easily crystallized due to a lack of metal binding sites on the surface of ferritin. The L-ferritin molecules in the crystals are linked to neighboring ferritin molecules by coordination of Cd2+ ions with aspartic acid and glutamine on the outer surface of ferritin. Thus, glutamine was introduced by replacement of Lys86 of human recombinant H-ferritin at the intermolecular contact position. This effort led to success in crystallization of H-ferritin in the presence of Ca2+ ions and provided a 3D structure of H-ferritin (Fig. 4a). | ||
| Fig. 4 Engineering protein crystallization by metal coordination. (a) Interactions of neighboring H-ferritin molecules. Ca2+ ions are coordinated with Asp and Gln on the surface of caged structure. The structure was taken from PDB code 1FHA. (b) Zn-mediated crystal packing of the T4L mutant. (c) Crystal structure of Zn-mediated MBPPhen2 derivative of cyt cb562 and the porous structures. (d) 2D sheet arrangement of the RIDC3 derivative of cyt cb562 in the presence of Zn2+ ions. Reproduced from ref. 32, 33 and 42 with permission for (b), (c) and (d) from Wiley, the Royal Society of Chemistry and Nature publishing group, respectively. | ||
Yeates and co-workers have reported a new crystallization methodology which combines the design of symmetric protein assemblies with the introduction of metal-binding sites on the surface of proteins (Fig. 4b).33 Histidine or cysteine residues were introduced on the surface of T4 lysozyme (T4L) and maltose-binding protein (MBP) to construct metal binding sites. Symmetric structures were then induced by binding of Cu2+, Ni2+ or Zn2+ ions and new lattice crystal structures of T4L and MBP were formed. The crystal structures show that oligomeric assembly structures are regulated by the number and position of metal binding sites and the characteristics of the metal ions. This methodology can be applied to crystallization of various asymmetric proteins and/or protein complexes.
Tezcan and co-workers have recently established a method for preparation of various protein supramolecules by design of metal-directed protein self-assemblies.14,42 The new method was applied to synthesize porous crystalline protein frameworks using engineered cytochrome cb562 and Ni2+ or Zn2+ ions (Fig. 4c).43 A variant of cytochrome cb562 was produced in which a Cys residue was introduced on the surface of cyt cb562 and two His residues were removed from the surface. The mutant was modified with a 1,10′-phenanthroline (Phen) moiety by reaction of the Cys residue and the iodoacetamido moiety of the Phen derivative. Crystals of the modified cyt cb562 (MBPPhen2) were obtained in the presence of Zn2+ or Ni2+ ions. The synthesized crystals have two highly ordered hexagonal channels 6 nm and 2 nm in length (Fig. 4c). It is expected that these porous frameworks will be used as solid materials. Ni and Tezcan have reported the construction of a tetrahedral supramolecular architecture in a rhombohedral crystal lattice by directed assembly of cytochrome cb562 with Zn2+ ions.40 A heme peptide fragment known as microperoxidase was immobilized by coordination of a His residue and hydrophobic interactions with exposed hydrophobic residues in the cavities of the cages, which have a diameter of 3.5 nm. This study suggests that crystallographic analyses enable the structural characterization of uncharacterized guest molecules.
Protein arrays such as 1D protein nanotubes, and 2D and 3D crystalline protein arrays have been controlled by using metal-directed cytochrome cb562 assembly (Fig. 4d).34 A mutant of cytochrome cb562 (RIDC3) was constructed which incorporates 12 surface mutations including 10 Rosetta-prescribed surface mutations and two metal-chelating bis-histidine mutations on the surface of cytochrome cb562. The 1D nanotubes, 2D sheets, and 3D crystals of RIDC3 are formed by coordination of Zn2+ ions. These 1D nanotubes and 2D sheets have high chemical stabilities due to self-assembly of RIDC3 monomers by metal coordination under high concentrations of polar organic solvents such as tetrahydrofuran (THF) and isopropanol (iPrOH), although the RIDC3 monomers denature in 30% THF and 50% iPrOH. The 1D and 2D assemblies also have thermal stability, up to 70 °C and 90 °C, respectively. Moreover, the Zn2+ ion-directed cytochrome cb562 assemblies were utilized as templates for growth of Pt0 nanoparticles on the surface of the assemblies with the aim of developing electron transfer systems.41
2.3. Protein crystallization via generation of intermolecular symmetry assembly by mutants
Another approaches to form symmetric protein assemblies for crystallization are disulfide linkages between the interfaces of protein monomers, and introduction of hydrophobic protein–protein interactions by site-directed mutagenesis.35,44 Yeates and co-workers reported an excellent strategy for formation of new protein crystal lattice structures by synthetic symmetrization of phage T4 lysozyme via disulfide bond formation (Fig. 5a and b).35 Single cysteine mutants of a pseudo-WT T4 lysozyme were prepared to produce a homodimer of lysozyme upon formation of a disulfide bond. Six novel crystal forms of Cys-introduced lysozyme, which cannot be obtained using a lysozyme monomer, were generated and characterized by X-ray diffraction analysis. In each of the structures, symmetric dimers formed by disulfide bonds were constructed in the crystals. This synthetic symmetrization approach using disulfide bonds is useful to obtain crystallization of otherwise unstable proteins as well as in the preparation of various lattice structures of the protein crystals in development of new materials. Quistgaard reported the preparation of a disulfide polymerized protein crystal by using a vDED coiled coil domain from BAP29 (Fig. 5c and d).44 The crystal structure shows the honeycomb-like pattern provided by disulfide formation across a crystal contact. The crystal has large solvent channels with dimensions of 75 × 90 Å, indicating that the system has significant potential for accumulation of functional molecules in the solvent channels.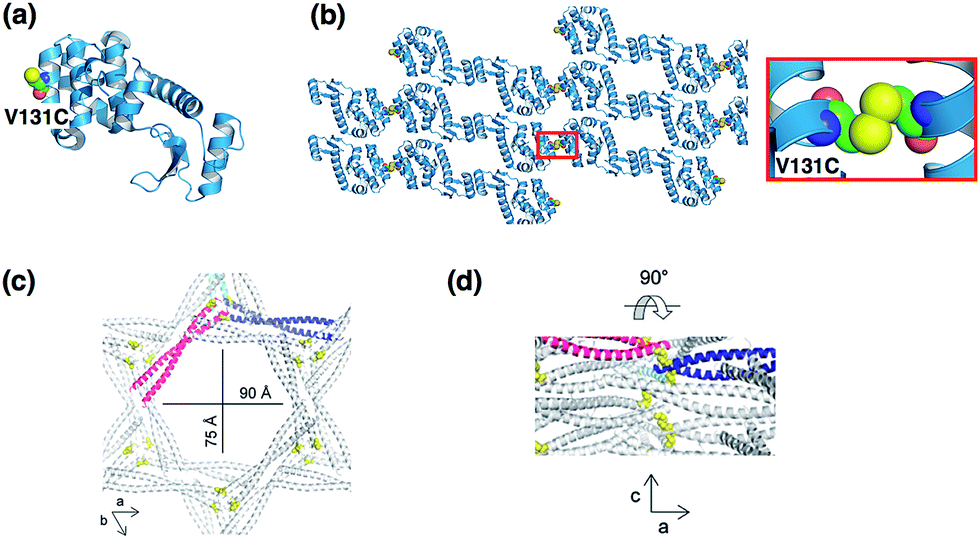 | ||
| Fig. 5 Protein crystallization via disulfide formation. (a) The position of the Cys residues of a T4 lysozyme mutant. (b) Crystal packing structure of V131C T4 lysozyme (space group: C2) and disulfide bond structure. The structure was taken from PDB code 2HUK. (c) The hexagonal solvent channel of BAP29. (d) Perpendicular view from within the channel. Reproduced from ref. 43 with permission for (c) and (d) from the Royal Society of Chemistry. | ||
Yamada et al. prepared several mutants of human RNase 1, whose crystal structure had not been reported at that time, to engineer a crystal lattice of RNase 1 by generation of hydrophobic protein–protein interactions.36 In the mutant 4L-RNase 1, four leucine residues were introduced to promote intermolecular symmetry by engineering of hydrophobic leucine-zipper like interactions. 4L-RNase 1 was successfully crystallized and the crystal structure shows that the introduced leucine residues generate new hydrophobic packing interfaces which induce the formation of crystals. This work suggests that intermolecular engineering by generation of leucine-zipper interfaces can produce symmetric assemblies which expand our ability to crystallize proteins.
2.4. 2D protein assembly using fusion technology
Yeates and co-workers constructed a large symmetric protein cage by self-assembly of fused subunits composed of two different proteins, which form natural dimer and trimer assemblies.45 However, the sizes of the cages were found to be heterogeneous due to association–dissociation equilibrium and formation of polymorphs. The original fusion proteins designs were improved by mutagenesis to prepare well-ordered homogeneous assemblies.46 The X-ray crystal structure of the assembly shows that the diameter of the cage is about 16 nm and the structure deviates from perfect symmetry by 8 Å overall.This fusion technology is available for 2D crystallization of proteins. Sinclair, Noble and co-workers have described the generation of a 2D crystalline lattice of proteins by using a D2 symmetric Streptag I/streptavidin assembly.37 A fusion of Streptag I to the D4 symmetric E. coli aminolevulinic acid dehydrogenase (ALAD) octamer was created. Then, the fusion proteins were reacted with streptavidin to generate a 2D crystal lattice structure by promotion of intermolecular interactions between Streptag I and streptavidin (Fig. 6). Transmission electron microscopy (TEM) and atomic force microscopy (AFM) of the assemblies show 2D crystalline lattice structures through association along a two-fold symmetry axis. It is expected that this method can be expanded to 3D crystals of proteins. Self-assembling 3D lattice structures were prepared, although without sufficient resolution for crystal structural analysis. These 3D structures were also found to be sensitive to heat and small molecule inhibitors.
 | ||
| Fig. 6 A 2D crystalline lattice structure containing ALAD and streptavidin/Streptag I assemblies. (a) The components of the assemblies, E. coli ALAD and streptavidin/Streptag I. These structures were obtained from PDB codes 1b4e and 1rst. (b) Lattice design of 2D crystalline assembly. (c) TEM images of the assemblies. Reproduced from ref. 36 with permission from Nature publishing group. | ||
2.5. Computational design of protein crystallization
The design of proteins that self-assemble into well-ordered protein assemblies is an important topic in the field of material sciences. Baker et al. have reported computational design of protein interfaces to generate highly ordered nano-sized protein architectures.47,48 Caged protein assemblies of 24-mers composed two distinct subunits were constructed based on computational design.48 This is a good example of how computational design can guide the construction of functional protein nanoarchitectures, which include two different subunits. Valuable applications are expected to arise from such efforts. Generation of artificial 3D protein crystals is attracting increasing attention because protein crystal structure analysis is a tool for structural characterization of biomacromolecules and protein crystals recently have been utilized as functional materials for applications in catalysis, development of therapeutic materials, and construction of molecular templates for synthesis of inorganic materials.3,4,18,24Computational design of proteins is applied to protein crystallization to regulate protein–protein interactions, such as hydrogen bond and hydrophobic interaction. Saven and co-workers computationally designed de novo three α-helix coiled coil proteins that form polar, layered 3D crystals with the P6 space group (Fig. 7).38 The coiled coil interactions of the proteins produced layered structures. Hydrogen bonds and hydrophobic interactions were designed for assembling layers to form 3D crystals. The X-ray crystal structure of the designed protein shows a honeycomb-like structure and hexameric channels. The root mean square deviation (rmsd) of the designed structure was about 1 Å relative to the computational models. This approach can be applied to form high-symmetry space groups of protein crystals and to control the content of the crystallization solvent, which can improve crystal quality and provide novel lattice structures of protein crystals.
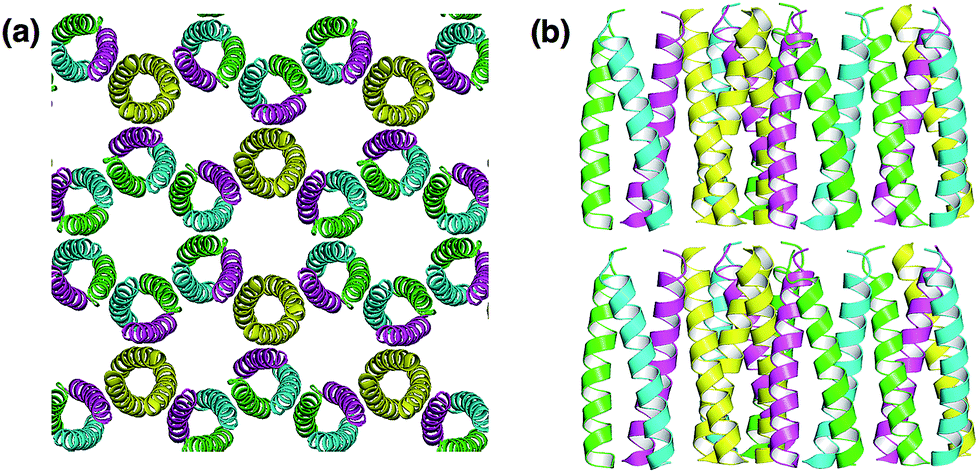 | ||
| Fig. 7 (a) Honeycomb-like P6 crystal lattice of a computationally-designed coiled-coil protein building block. (b) The layer structures were stacked by hydrogen bond interactions between layers. The structure was taken from PDB code 4DAC. | ||
3. Protein crystals as porous materials
The properties of protein crystals are comparable to typical porous materials such as zeolites and mesoporous silica. As such, protein crystals offer a wide range of pore sizes (20–100 Å), high porosity (0.5–0.8), and large pore surface areas (800–2000 m2 g−1).18,19 In contrast, the pore size and pore surface area are 2–10 Å and 500–2000 m2 g−1 for zeolites,49,50 and 20–300 Å, and 700–1500 m2 g−1 for mesoporous silica.51 Compared to the conventional porous materials, the advantages of protein crystals as porous materials are: (1) functional groups of side chains of amino acid residues are periodically aligned on the porous surface of crystals, (2) metal ions, metal complexes and functional molecules can be periodically accumulated inside of porous space by coordination and chemical modification, (3) porous space are constructed by inherently chiral environments of protein molecules. However, most protein crystals are very fragile compared to the conventional porous materials, because the protein crystals are prone to dissolution when the crystals are soaked in the solution other than the solution of crystallization conditions. Therefore, cross-linked enzyme crystals (CLECs) and cross-linked protein crystals (CLPCs) have been developed to strengthen the crystals to utilize as porous materials.3.1. Cross-linked enzyme crystals
CLECs and CLPCs have been utilized as biomaterials for applications in separation processes, heterogeneous catalysis, and drug delivery systems because CLECs and CLPCs are extremely stable against mechanical disruption and reactions conducted both in water and organic solvents.18Margolin et al. used cross-linked Candida rugosa lipase (CRL) crystals for enantioselective hydrolysis of chiral esters.52 CRL crystals cross-linked with glutaraldehyde are highly stable in aqueous media and organic solvents and can be recovered for recycling. The CRL-CLECs have higher enantioselectivity in hydrolysis reactions than crude commercial CRL because the pure CRL is obtained by crystallization. The same researchers have also reported that CLPCs can be used as porous materials for separating molecules.19 Thermolysin-CLPCs and human serum albumin (HSA)-CLPCs provide valuable separation materials as carriers for column chromatography by simultaneously providing three separation mechanisms; size exclusion, adsorption, and chirality. These initial excellent results indicate that the CLECs and CLPCs function as bioorganic porous materials, which are comparable to zeolites and mesoporous silica.
Protein crystals capture and deposit metal ions and metal complexes within solvent channels when the crystals are soaked in solutions containing the metal ions or metal complexes because the amino acid side chains responsible for coordinating the metal ions are periodically aligned on the surface of solvent channels of the crystal lattices.53–55 Therefore, protein crystals have been functionalized for applications in preparation of inorganic materials, catalysis, and accumulation of functional compounds.
3.2. Preparation of inorganic materials
Solvent channels of protein crystals have been utilized for preparation of inorganic nanoparticles by coordination interaction with amino acid residues exposed on the channel surface.Colvin et al. reported utilization of protein crystals as templates for preparation of inorganic materials.56 The crystal of cowpea mosaic virus (CPMV) was cross-linked by glutaraldehyde and then soaked in a buffer solution containing Pd2+ ions. The Pd2+ ions were found to accumulate in the solvent channels of CPMV crystals by coordination to amino acid residues. Next, the crystals accumulating Pd2+ ions were exposed to a solution containing Pt2+ ions and sodium hypophosphite. The coordinated Pd2+ ions were reduced by the hypophosphite. The reduced Pd0 catalyzes the electrolytic deposition of Pt2+ in the channels of the crystals. A scanning electron microscope (SEM) image and energy-dispersive X-ray (EDX) analysis of a metal-filled CPMV crystal each provide evidence for homogeneous distribution of Pd and Pt metal nanoparticles (Fig. 8). The electron transmission microscopy (TEM) image of the crystal indicated that the resulting metallic nanoparticles are highly aligned within the solvent cavities of the crystal array. This study suggests that solvent channels of protein crystals can be utilized as templates for synthesis of metal nanoparticles and that this method can be applied to various protein crystals.
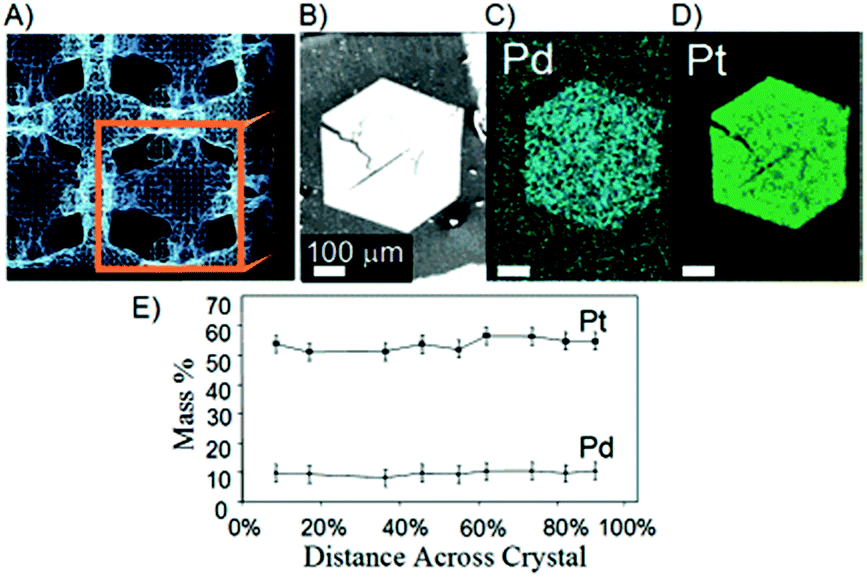 | ||
| Fig. 8 (a) Porous spaces between viruses within a crystal structure generated by XRD data. (b) SEM image of a cross-linked CPMV crystal after metallization. X-ray backscattering of (c) the palladium and (d) platinum distribution in the crystal. (e) EDX analysis of the metal distribution in the same crystal. Reproduced from ref. 52 with permission from American Chemical Society. | ||
The hen egg white lysozyme (HEWL) crystal is an excellent candidate for use as a molecular template for synthesis of metal nanoparticles because it has a well-characterized capacity for accumulation of various metal ions and metal complexes by coordination of amino acid residues in the solvent channels of the crystals.53–55
These crystals can form various polymorphic structures, including tetragonal, orthorhombic and monoclinic structures (T-, O-, and M-HEWL), according to the crystallization conditions employed. HEWL is easily and abundantly crystallized. Therefore, there have been several reports investigating the use of HEWL crystals as templates for synthesis of metallic nano-composites.57–59
Mann et al. have synthesized metallic nano-filaments within the 1D solvent channels of the tetragonal HEWL crystal (P43212) (T-HEWL), which has regular zigzag solvent channels with a pore diameter range of 1 to 2.2 nm.57 After glutaraldehyde cross-linking of T-HEWL (CL-T-HEWL), Au3+ or Ag+ ions were accumulated in the solvent channels and these metal ions were reduced by chemical reduction or photoreduction methods to synthesize metallic nano-rods in the solvent channels of the crystals (Fig. 9). The metallic rods were disposed in a well-ordered array along the solvent channels of lysozyme crystals. Diffuse reflectance UV/Vis spectra of metal-filled CL-T-HEWL indicates the transverse (412 and 583 nm for Ag and Au, respectively) and longitudinal (505 and 684 nm for Ag and Au, respectively) plasmon resonance peaks. Furthermore, polypyrrole (PPy) has been synthesized in the crystals. CL-T-HEWL crystals were soaked in the aqueous solution containing ammonium persulfate (APS), an oxidant for pyrrole.60 Then APS-CL-T-HEWL was exposed to pyrrole vapor for synthesis of PPy in the solvent channels of the crystals. The resulting hybrid materials were found to be insoluble in water and organic solvent, electrically conductive, and mechanically plastic compared to unmodified CL-T-HEWL.
 | ||
| Fig. 9 (a) Optical and SEM micrographs for chemically-reduced Au containing cross-linked lysozyme crystals. (b) TEM image of Au-filled lysozyme crystals and corresponding EDX analysis and electron diffraction data. Reproduced from ref. 53 with permission from Wiley-VCH. | ||
Lu and co-workers have reported the mechanism of growth of gold nanoparticles (Au NPs) within a single HEWL crystal.58 Time-dependent formation of Au NPs in the protein assemblies was observed using a HEWL crystal. The HEWL crystal accumulating ClAuS(CH2CH2OH)2 (Au+) is an appropriate template for use in elucidating the processes of Au NP formation because the kinetic formation of Au NPs promoted by auto-reduction of Au+ in the crystal is slow. The growth and distribution of Au NPs within the crystals were monitored with transmission electron microscopy (TEM) and the interactions of gold ions with amino acid residues were evaluated by X-ray crystal structural analyses (Fig. 10). The time-dependent TEM images showed that Au NPs gradually grew within the crystals from 2.9 nm after 1.5 day of growth to 16.9 nm after 10 days (Fig. 10b). X-ray crystal structures indicate that Au+ initially binds to His15 in the solvent channel of the lysozyme crystal. Then Au+ dissociates from His15 and disproportionates into Au0 and Au3+ (Fig. 10c). During the process of Au NP formation, free Au+ is continuously incorporated into the crystals and disproportionates into Au0 and Au3+, and Au0 which then accumulates into the Au NPs. This work could provide a new method for elucidating the mechanisms of biomineralization processes and metal nanocluster formation. It was also reported that the lysozyme crystals containing Au NPs could be used as catalysts for reduction of p-nitrophenol by NaBH4.61 The catalytic activities of the Au NPs in the HEWL crystal could be regulated by the size of Au NPs, which are precisely controlled by HEWL crystals.
 | ||
| Fig. 10 (a) Optical images of single crystals of lysozyme grown in the presence of Au(I) on different days of growth. (b) Corresponding TEM images at high magnification. (c) X-ray crystal structures of lysozyme from single crystals at the first, second, third and 90th days of growth. Reproduced from ref. 54 with permission from Nature publishing group. | ||
Ueno and co-workers reported the synthesis of magnetic bimetallic CoPt NPs within HEWL crystals and succeeded in controlling the magnetic properties of the CoPt NPs by using different crystal systems of HEWL crystals (Fig. 11).59 The X-ray crystal analyses of a cross-linked orthorhombic HEWL crystal (CL-O-HEWL) soaked in a buffer solution containing Co2+ ions show that Co2+ ions bind to Asp52. Pt2+·CL-O-HEWL has five binding sites for Pt2+ in the vicinity of Lys1, Cys6, His15, and Asp119. The results indicate that Co2+ and Pt2+ ions coordinate to different sites in the same channels of HEWL. CoPt NPs were synthesized by reduction of Co2+ and Pt2+ accumulated in the solvent channels of CL-O-HEWL, CL-T-HEWL, and CL-M-HEWL crystals with sodium borohydride. Transmission electron microscope (TEM) images show that the size of the CoPt NPs synthesized in CL-O-HEWL, CL-T-HEWL, and CL-M-HEWL are 1.9 ± 0.3, 1.7 ± 0.4, and 1.4 ± 0.3 nm, respectively. These sizes are commensurate with the space of porous solvent channels of HEWL crystals. Interestingly, these NPs are aligned in solvent channels of the CL-HEWL crystals, suggesting that the NPs were synthesized in the largest solvent channels of the crystals (Fig. 11e–g). X-ray fluorescence measurements of CoPt NPs in CL-O-HEWL, CL-T-HEWL, and CL-M-HEWL show that the ratios of Co and Pt atoms in CoPt NPs are 7.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :92.3, 3.8:96.2, and 6.3:93.7, respectively. Pre-organization of Co2+ and Pt2+ ions in the solvent channels of HEWL crystals would affect the compositions of CoPt NPs. The magnetic properties of CoPt·CL-HEWLs were characterized by a superconducting quantum interference device (SQUID) magnetometer. CoPt·CL-O-HEWL has high a coercivity value (4600 Oe) compared to those of CoPt·CL-T-HEWL and CoPt·CL-M-HEWL (1600 and 2900 Oe, respectively). The order of the coercivity of the CoPt-NPs in the HEWL crystals follows the composition ratio of Co of CoPt NPs because the coercivity of CoPt NP increases with Co content up to 1:3 of the Co to Pt ratio. These results suggest that the properties of inorganic metal nanoparticles can be controlled by pre-organization of different metal ions, which are dominated by lattice structures of protein crystals.
:92.3, 3.8:96.2, and 6.3:93.7, respectively. Pre-organization of Co2+ and Pt2+ ions in the solvent channels of HEWL crystals would affect the compositions of CoPt NPs. The magnetic properties of CoPt·CL-HEWLs were characterized by a superconducting quantum interference device (SQUID) magnetometer. CoPt·CL-O-HEWL has high a coercivity value (4600 Oe) compared to those of CoPt·CL-T-HEWL and CoPt·CL-M-HEWL (1600 and 2900 Oe, respectively). The order of the coercivity of the CoPt-NPs in the HEWL crystals follows the composition ratio of Co of CoPt NPs because the coercivity of CoPt NP increases with Co content up to 1:3 of the Co to Pt ratio. These results suggest that the properties of inorganic metal nanoparticles can be controlled by pre-organization of different metal ions, which are dominated by lattice structures of protein crystals.
 | ||
| Fig. 11 (a) Schematic drawings illustrating the preparation of CoPt-NPs in a solvent channel of a HEWL crystal. Crystal lattice structures and the major solvent channels of (b) O-HEWL, (c) T-HEWL, and (d) M-HEWL taken from the PDB codes of 1BGI, 103L, and 5LYM, respectively. TEM images and particle size distribution of (e) CoPt·CL-O-HEWL, (f) CoPt·CL-T-HEWL, and (g) CoPt·CL-M-HEWL. The black dots are CoPt NPs and the white dotted line (insets) provide a guide. | ||
3.3. Functionalization of porous protein crystals
Functionalization of the solvent channels of protein crystals has recently been attractive because the crystals can provide nano-sized environments to immobilize metal ions and metal complexes for applications, such as asymmetric solid catalysts, extracellular matrices and electron transfer systems.24,62–64Post-engineering of porous protein crystals to convert solvent channels into catalytic vessels by incorporation of organometallic complexes was conducted by Ueno et al.62 Ru(benzene)Cl2 complexes are immobilized in the solvent channels of CL-HEWL crystals by coordination of His15 (Fig. 12). The Ru immobilized HEWL crystals catalyze the enantioselective transfer hydrogenation of acetophenone derivatives. Ru(benzene)Cl2·CL-T-HEWL (Ru(benzene)·CL-T-HEWL) shows the highest catalytic activity for propiophenone with 53% conversion. A buffer solution containing catalytic Ru complexes with HEWL monomers was found to promote conversion of transfer hydrogenation at rates 4–8 fold lower than those of Ru(benzene)·CL-T-HEWL. The buffer solution of Ru complexes without HEWL monomers was found to have no catalytic activity under the same conditions. These results indicate that immobilization of Ru complexes in the solvent channels of lysozyme crystals promotes hydrogenation activity. Ru(benzene)Cl2·CL-O-HEWL (Ru(benzene)·CL-O-HEWL) exhibits the highest ee value for isobutyrophenone with 36% ee of the S enantiomer. Ru(benzene)·CL-O-HEWL has higher enantioselectivity than Ru(benzene)·CL-T-HEWL for all the products of transfer hydrogenation. The hydrogenation enantioselectivity and configurations were found to be controlled by interactions of substrates with the Ru complexes within the crystals, which are affected by the crystal lattice structures and hydrogen bonding networks of the crystals.
 | ||
| Fig. 12 (a) Schematic representation of the preparation of coordinated Ru(benzene)Cl2·His in the solvent channel of a porous HEWL crystal. Coordination structures in a lattice structure of (b) Ru(benzene)·CL-T-HEWL and (d) Ru(benzene)·CL-O-HEWL, and active-site structures with hydrogen-bonding networks (c: Ru(benzene)·CL-T-HEWL, e: Ru(benzene)·CL-O-HEWL). | ||
CL-T-HEWL has been engineered as an extracellular matrix for storage and release of CO gas by immobilization of ruthenium carbonyl organometallic complexes.64 Ru carbonyl complexes were immobilized by coordination of histidine and aspartic acid residues in the solvent channels of the CL-T-HEWL. The Ru-modified CL-T-HEWL (RuCO·CL-T-HEWL) releases CO under physiological conditions, although a composite of HEWL with Ru carbonyl complexes dissolved in the buffer solution do not release CO. When a RuCO·CL-T-HEWL was used as an extracellular scaffold, nuclear factor kappa B (NF-κB) activity in living cells was found to increase by effective delivery of CO due to adhesion of crystals with cells and slower release of CO from modified crystals. This work suggests that protein crystals incorporating metal complexes could be used as extracellular matrices for delivery of signaling molecules such as gases and proteins to living cells.
Accumulation of metal ions on the protein surface is a crucial step in the process of forming inorganic materials and small metal-clusters. The mechanism of accumulation of metal ions on the surface of proteins was elucidated using X-ray crystal structural analysis of a series of HEWL crystals containing Rh ions (Fig. 13).65 T-HEWL crystals were soaked in the buffer solutions containing different concentrations of RhCl3 (0, 1, 5, 10, 30 and 100 mM) at 20 °C for 2 days. X-ray crystal structures of these Rh/T-HEWL crystals show that Rh ions were deposited at the five binding sites on the surface of solvent channels of T-HEWL by coordination of amino acid residues or by hydrogen bonding networks of Rh coordinated water molecules with amino acid residues. At each of the binding sites, metal ions are accumulated by the action of highly cooperative dynamics of side chains of amino acid residues and hydrogen bonds. At site B, two Rh3+ ions are accumulated, accompanied by large conformational changes of side chains of Asp 18 and Asn 19 induced by disruption of hydrogen bonds in the vicinity of Asn19 (Fig. 13c). These results suggest that rearrangement of hydrogen bond networks and conformational changes of amino acid residues induce the process of metal accumulation on the protein surface. These interactions promote the natural metal accumulation reactions such as biomineralization and formation of metal clusters.
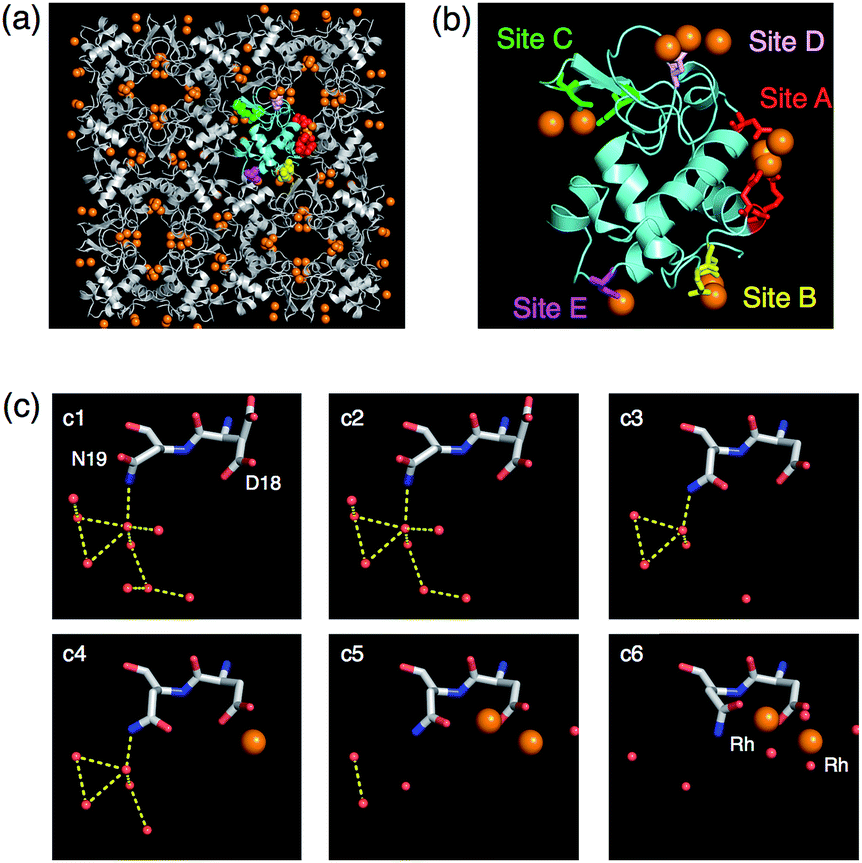 | ||
| Fig. 13 The crystal lattice and molecular structures of 100-Rh·T-HEWL (a and b). The monomer is indicated by the cyan structure in (a) and (b). (c) Snapshot analyses of Rh ion binding to site B. Each structure obtained at a different concentration of RhCl3 is indicated by c1, c2, c3, c4, c5, and c6 representing 0, 1, 5, 10, 30, and 100 mM RhCl3, respectively, in sodium acetate buffer at pH 4.5. The Rh and O atoms are indicated by orange and red spheres, respectively. The yellow dashed lines indicate hydrogen bonds. The side chains of Asp18 in c1 and c2 are dual conformers. | ||
The accumulation of functional molecules such as fluorescein, eosin and a Ru(bpy)3 complex into the hexagonal solvent channels of porous myoglobin crystals was demonstrated.66 The crystal of recombinant sperm whale myoglobin (Mb) belongs to space group P6 and has hexagonal pores consisting of the building blocks of six Mb molecules, which form a pore with a diameter of 40 Å.67 The cysteine residues of these functional molecules were modified by thiol-maleimide chemistry. Then the modified Mb was crystallized to prepare a crystal retaining P6 lattice structure. The resulting crystals show that the functional molecules are efficiently immobilized with well-ordered alignment in the hexagonal channels of the Mb crystals.
An artificial photoinduced electron transfer system was constructed by accumulating redox cofactors in a single crystal of Mb (Fig. 14).63 Electron transfer mediated by methyl viologen (MV) occurs between the Zn porphyrin (ZnP) and the Ru3O cluster, which are precisely immobilized in the crystals. This modified crystal provides a long half-life to the charge-separated state which is 2800-fold longer than that of the previously reported system in organic solution because of site-specific immobilization of the ZnP, the Ru3O cluster and MV in the crystal of Mb (Fig. 14). The ZnP, the Ru3O cluster and MV are accumulated in the three dimensional space of myoglobin by cofactor replacement with heme, coordination of His residues and diffusion in the solvent channel, respectively. This report describes the advantage of using protein crystals to generate an artificial electron transfer system with a long-lived charged separated state by providing a dense array of redox cofactors in the crystals.
 | ||
| Fig. 14 (a) Construction of a photoinduced electron transfer system in Mb crystals. (b) Dense array of Ru3O and ZnP in the ZnMb/Ru3O crystal. Reproduced from ref. 59 with permission from Wiley-VCH. | ||
4. Conclusions
This review article summarizes the impressive progress of protein crystals in materials science. Several protein engineering approaches have been developed to generate novel crystal lattices of protein assemblies. It is important to generate symmetric protein oligomerization by design of interfaces between protein monomers, which facilitate protein crystallization. Solvent channels of protein crystals are capable of accumulating exogenous molecules by metal coordination and chemical modification with the amino acid residues on the surface of solvent channels without disruption of the crystal lattices. Protein crystals, which immobilize metal complexes and metal ions in the solvent channels, can function as catalysts with high reactivity, selectively, and recyclability, and can be used to prepare inorganic nanoparticles with magnetic and catalytic properties. The further challenges of protein crystalline materials are not only to functionalize protein crystals with novel crystal lattice by design of protein interfaces but also to establish large scale or more conventional preparation systems of tailored protein crystals. We believe that engineering of protein crystals can provide remarkable advances in the fields of both material sciences and structural biology.Acknowledgements
Parts of this work were supported by the Funding Program for Next Generation World-Leading Researchers (for T. U.) and Grant-in Aid for Scientific Research (for S. A.) from Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- M. Uchida, M. T. Klem, M. Allen, P. Suci, M. Flenniken, E. Gillitzer, Z. Varpness, L. O. Liepold, M. Young and T. Douglas, Adv. Mater., 2007, 19, 1025–1042 CrossRef CAS.
- S. Howorka, Curr. Opin. Biotechnol., 2011, 22, 485–491 CrossRef CAS PubMed.
- N. J. M. Sanghamitra and T. Ueno, Chem. Commun., 2013, 49, 4114–4126 RSC.
- T. Ueno, H. Tabe and Y. Tanaka, Chem.–Asian J., 2013, 8, 1646–1660 CrossRef CAS PubMed.
- T. Ueno, M. Suzuki, T. Goto, T. Matsumoto, K. Nagayama and Y. Watanabe, Angew. Chem., Int. Ed., 2004, 43, 2527–2530 CrossRef CAS PubMed.
- M. Suzuki, M. Abe, T. Ueno, S. Abe, T. Goto, Y. Toda, T. Akita, Y. Yamadae and Y. Watanabe, Chem. Commun., 2009, 4871–4873 RSC.
- S. Abe, J. Niemeyer, M. Abe, Y. Takezawa, T. Ueno, T. Hikage, G. Erker and Y. Watanabe, J. Am. Chem. Soc., 2008, 130, 10512–10514 CrossRef CAS PubMed.
- S. Abe, K. Hirata, T. Ueno, K. Morino, N. Shimizu, M. Yamamoto, M. Takata, E. Yashima and Y. Watanabe, J. Am. Chem. Soc., 2009, 131, 6958–6960 CrossRef CAS PubMed.
- J. Lucon, S. Qazi, M. Uchida, G. J. Bedwell, B. LaFrance, P. E. Prevelige and T. Douglas, Nat. Chem., 2012, 4, 781–788 CrossRef CAS PubMed.
- Coordination Chemistry in Protein Cages: Principles, Design, and Applications, ed. T. Ueno and Y. Watanabe, John Wiley & Sons, New Jersey, 2013 Search PubMed.
- R. A. Miller, A. D. Presley and M. B. Francis, J. Am. Chem. Soc., 2007, 129, 3104–3109 CrossRef CAS PubMed.
- N. Carette, H. Engelkamp, E. Akpa, S. J. Pierre, N. R. Cameron, P. C. M. Christianen, J. C. Maan, J. C. Thies, R. Weberskirch, A. E. Rowan, R. J. M. Nolte, T. Michon and J. C. M. Van Hest, Nat. Nanotechnol., 2007, 2, 226–229 CrossRef CAS PubMed.
- D. Ghosh, Y. Lee, S. Thomas, A. G. Kohli, D. S. Yun, A. M. Belcher and K. A. Kelly, Nat. Nanotechnol., 2012, 7, 677–682 CrossRef CAS PubMed.
- P. A. Sontz, W. J. Song and F. A. Tezcan, Curr. Opin. Chem. Biol., 2014, 19, 42–49 CrossRef CAS PubMed.
- Y. T. Lai, N. P. King and T. O. Yeates, Trends Cell Biol., 2012, 22, 653–661 CrossRef CAS PubMed.
- Q. Luo, Z. Y. Dong, C. X. Hou and J. Q. Liu, Chem. Commun., 2014, 50, 9997–10007 RSC.
- J. C. Sinclair, Curr. Opin. Chem. Biol., 2013, 17, 946–951 CrossRef CAS PubMed.
- A. L. Margolin and M. A. Navia, Angew. Chem., Int. Ed., 2001, 40, 2205–2222 CrossRef.
- L. Z. Vilenchik, J. P. Griffith, N. St Clair, M. A. Navia and A. L. Margolin, J. Am. Chem. Soc., 1998, 120, 4290–4294 CrossRef CAS.
- N. Ilk, E. M. Egelseer and U. B. Sleytr, Curr. Opin. Biotechnol., 2011, 22, 824–831 CrossRef CAS PubMed.
- D. Moll, C. Huber, B. Schlegel, D. Pum, U. B. Sleytr and M. Sara, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14646–14651 CrossRef CAS PubMed.
- H. Tschiggerl, A. Breitwieser, G. de Roo, T. Verwoerd, C. Schaffer and U. B. Sleytr, J. Biotechnol., 2008, 133, 403–411 CrossRef CAS PubMed.
- E. Baranova, R. Fronzes, A. Garcia-Pino, N. Van Gerven, D. Papapostolou, G. Pehau-Arnaudet, E. Pardon, J. Steyaert, S. Howorka and H. Remaut, Nature, 2012, 487, 119–122 CAS.
- T. Ueno, Chem.–Eur. J., 2013, 19, 9096–9102 CrossRef CAS PubMed.
- C. Geng and P. J. Paukstelis, J. Am. Chem. Soc., 2014, 136, 7817–7820 CrossRef CAS PubMed.
- J. P. Zheng, J. J. Birktoft, Y. Chen, T. Wang, R. J. Sha, P. E. Constantinou, S. L. Ginell, C. D. Mao and N. C. Seeman, Nature, 2009, 461, 74–77 CrossRef CAS PubMed.
- T. Sawada, A. Matsumoto and M. Fujita, Angew. Chem., Int. Ed., 2014, 53, 7228–7232 CrossRef CAS PubMed.
- A. Mantion, L. Massuger, P. Rabu, C. Palivan, L. B. McCusker and A. Taubert, J. Am. Chem. Soc., 2008, 130, 2517–2526 CrossRef CAS PubMed.
- A. McPherson and J. A. Gavira, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2014, 70, 2–20 CrossRef CAS PubMed.
- N. Dotan, D. Arad, F. Frolow and A. Freeman, Angew. Chem., Int. Ed., 1999, 38, 2363–2366 CrossRef CAS.
- F. Sakai, G. Yang, M. S. Weiss, Y. J. Liu, G. S. Chen and M. Jiang, Nat. Commun., 2014, 5, 4634 CrossRef PubMed.
- D. M. Lawson, P. J. Artymiuk, S. J. Yewdall, J. M. A. Smith, J. C. Livingstone, A. Treffry, A. Luzzago, S. Levi, P. Arosio, G. Cesareni, C. D. Thomas, W. V. Shaw and P. M. Harrison, Nature, 1991, 349, 541–544 CrossRef CAS PubMed.
- A. Laganowsky, M. L. Zhao, A. B. Soriaga, M. R. Sawaya, D. Cascio and T. O. Yeates, Protein Sci., 2011, 20, 1876–1890 CrossRef CAS PubMed.
- J. D. Brodin, X. I. Ambroggio, C. Y. Tang, K. N. Parent, T. S. Baker and F. A. Tezcan, Nat. Chem., 2012, 4, 375–382 CrossRef CAS PubMed.
- D. R. Banatao, D. Cascio, C. S. Crowley, M. R. Fleissner, H. L. Tienson and T. O. Yeates, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16230–16235 CrossRef CAS PubMed.
- H. Yamada, T. Tamada, M. Kosaka, K. Miyata, S. Fujiki, M. Tano, M. Moriya, M. Yamanishi, E. Honjo, H. Tada, T. Ino, H. Yamaguchi, J. Futami, M. Seno, T. Nomoto, T. Hirata, M. Yoshimura and R. Kuroki, Protein Sci., 2007, 16, 1389–1397 CrossRef CAS PubMed.
- J. C. Sinclair, K. M. Davies, C. Venien-Bryan and M. E. M. Noble, Nat. Nanotechnol., 2011, 6, 558–562 CrossRef CAS PubMed.
- C. J. Lanci, C. M. MacDermaid, S. G. Kang, R. Acharya, B. North, X. Yang, X. J. Qiu, W. F. DeGrado and J. G. Saven, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7304–7309 CrossRef CAS PubMed.
- S. W. Wukovitz and T. O. Yeates, Nat. Struct. Biol., 1995, 2, 1062–1067 CrossRef CAS.
- T. W. Ni and F. A. Tezcan, Angew. Chem., Int. Ed., 2010, 49, 7014–7018 CrossRef CAS PubMed.
- J. D. Brodin, J. R. Carr, P. A. Sontz and F. A. Tezcan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2897–2902 CrossRef CAS PubMed.
- E. N. Salgado, R. J. Radford and F. A. Tezcan, Acc. Chem. Res., 2010, 43, 661–672 CrossRef CAS PubMed.
- R. J. Radford, M. Lawrenz, P. C. Nguyen, J. A. McCammon and F. A. Tezcan, Chem. Commun., 2011, 47, 313–315 RSC.
- E. M. Quistgaard, Chem. Commun., 2014, 50, 14995–14997 RSC.
- J. E. Padilla, C. Colovos and T. O. Yeates, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2217–2221 CrossRef CAS PubMed.
- Y. T. Lai, D. Cascio and T. O. Yeates, Science, 2012, 336, 1129 CrossRef CAS PubMed.
- N. P. King, W. Sheffler, M. R. Sawaya, B. S. Vollmar, J. P. Sumida, I. Andre, T. Gonen, T. O. Yeates and D. Baker, Science, 2012, 336, 1171–1174 CrossRef CAS PubMed.
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef CAS PubMed.
- J. M. Garces, Adv. Mater., 1996, 8, 434–437 CrossRef CAS.
- M. E. Davis, A. Katz and W. R. Ahmad, Chem. Mater., 1996, 8, 1820–1839 CrossRef CAS.
- B. G. Trewyn, I. I. Slowing, S. Giri, H. T. Chen and V. S. Y. Lin, Acc. Chem. Res., 2007, 40, 846–853 CrossRef CAS PubMed.
- J. J. Lalonde, C. Govardhan, N. Khalaf, A. G. Martinez, K. Visuri and A. L. Margolin, J. Am. Chem. Soc., 1995, 117, 6845–6852 CrossRef CAS.
- M. Razavet, V. Artero, C. Cavazza, Y. Oudart, C. Lebrun, J. C. Fontecilla-Camps and M. Fontecave, Chem. Commun., 2007, 2805–2807 RSC.
- I. W. McNae, K. Fishburne, A. Habtemariam, T. M. Hunter, M. Melchart, F. Y. Wang, M. D. Walkinshaw and P. J. Sadler, Chem. Commun., 2004, 1786–1787 RSC.
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernardes, C. C. Romao and M. J. Romao, J. Am. Chem. Soc., 2011, 133, 1192–1195 CrossRef CAS PubMed.
- J. C. Falkner, M. E. Turner, J. K. Bosworth, T. J. Trentler, J. E. Johnson, T. W. Lin and V. L. Colvin, J. Am. Chem. Soc., 2005, 127, 5274–5275 CrossRef CAS PubMed.
- M. Guli, E. M. Lambert, M. Li and S. Mann, Angew. Chem., Int. Ed., 2010, 49, 520–523 CrossRef CAS PubMed.
- H. Wei, Z. D. Wang, J. O. Zhang, S. House, Y. G. Gao, L. M. Yang, H. Robinson, L. H. Tan, H. Xing, C. J. Hou, I. M. Robertson, J. M. Zuo and Y. Lu, Nat. Nanotechnol., 2011, 6, 92–96 CrossRef PubMed.
- S. Abe, M. Tsujimoto, K. Yoneda, M. Ohba, T. Hikage, M. Takano, S. Kitagawa and T. Ueno, Small, 2012, 8, 1314–1319 CrossRef CAS PubMed.
- M. W. England, E. M. Lambert, M. Li, L. Turyanska, A. J. Patil and S. Mann, Nanoscale, 2012, 4, 6710–6713 RSC.
- H. Wei and Y. Lu, Chem.–Asian J., 2012, 7, 680–683 CrossRef CAS PubMed.
- H. Tabe, S. Abe, T. Hikage, S. Kitagawa and T. Ueno, Chem.–Asian J., 2014, 9, 1373–1378 CrossRef CAS PubMed.
- T. Koshiyama, M. Shirai, T. Hikage, H. Tabe, K. Tanaka, S. Kitagawa and T. Ueno, Angew. Chem., Int. Ed., 2011, 50, 4849–4852 CrossRef CAS PubMed.
- H. Tabe, K. Fujita, S. Abe, M. Tsujimoto, T. Kuchimaru, S. Kizaka-Kondoh, M. Takano, S. Kitagawa and T. Ueno, Inorg. Chem., 2015, 54, 215–220 CrossRef CAS PubMed.
- T. Ueno, S. Abe, T. Koshiyama, T. Ohki, T. Hikage and Y. Watanabe, Chem.–Eur. J., 2010, 16, 2730–2740 CrossRef CAS PubMed.
- T. Koshiyama, N. Kawaba, T. Hikage, M. Shirai, Y. Miura, C. Y. Huang, K. Tanaka, Y. Watanabe and T. Ueno, Bioconjugate Chem., 2010, 21, 264–269 CrossRef CAS PubMed.
- G. N. Phillips, R. M. Arduini, B. A. Springer and S. G. Sligar, Proteins: Struct., Funct., Genet., 1990, 7, 358–365 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |